All published articles of this journal are available on ScienceDirect.

The Toll-like Receptor 2/6 Ligand MALP-2 Reduces the Viability of Mycobacterium tuberculosis in Murine Macrophages
Authors Info & Affiliations
Abstract
Toll-like receptors (TLRs) sense conserved structures of pathogens and influence macrophage functions. Here we investigated the impact of TLR signaling on the modulation of macrophage defense mechanisms against infection of Mycobacterium tuberculosis (MTB), the causative agent of tuberculosis. We found that a synthetic derivative of the TLR2/6 agonist MALP-2 and the potent TLR4 agonist lipopolysaccharide inhibited the intracellular growth of MTB in murine macrophages. Likely the microbicidal effect was mediated by production of nitric oxide while it is still unclear the role played by release of TNF-α , IL-6, MIP-1β and IL-10. These results suggest that the activation of microbicidal defense via TLR ligands is an appealing target for the establishment on immune intervention against tuberculosis.
INTRODUCTION
A series of pattern recognition receptors such as Toll-like receptors (TLRs) sense conserved structures of pathogens and activate dendritic cells (DC) and macrophages. Signals via TLRs enhance DC antigen-presentation and favour the development of B and T cell immunity during vaccination [1], thus several TLR ligands are under investigation as novel candidate vaccine adjuvant. On macrophages, triggering of TLRs promote the release of pro-inflammatory cytokines, chemokines, nitric oxide (NO), and prostaglandins [2, 3], which in turn can affect microbicidal defence mechanisms. The impact of TLR signalling on macrophage pathogen growth control is particularly relevant for those intracellular pathogens, such as Mycobacterium tuberculosis (MTB), which survive and grow primarily within host macrophages. MTB is the causative agent of tuberculosis (TB), a still dreadful infectious disease which causes of almost 2 million deaths per year [4]. MTB or its cell wall lipids, bind to several TLRs expressed on macrophages. Mycobacterial ligands for TLR2 or the heterodimers TLR2/1 and TLR2/6 include the mycobacterial p19 lipoprotein arabinosecapped Lipoarabinomannan (LAM), PIM, lipomannan and trehalose dimycolate; while for TLR9 the CpG-containing DNA, and for TLR4, a heat-sensitive but still unidentified mycobacterial component have been established as ligands [5, 6]. Engagement of macrophage TLRs by mycobacterial ligands promotes the expression of pro-inflammatory mediators (IL-12, IL-18, TNF-α, MCP-1, and MIP1α) and antimycobacterial effectors (Irgm-1/LRG47, cathelicidin, NO and other reactive nitrogen intermediates), and also induces phagosome maturation [5, 7, 8, 9]. In addition, signalling through MyD88, the adaptor molecule necessary for all TLR (apart from TLR3), is essential for protection against MTB infection in mice [10]. However, MTB has developed several immune-evasion mechanisms inside the macrophage, often through TLR ligands, such as the19 kDa mycobacterial lipoprotein, which dampen the innate immune response and allow MTB to survive also in activated macrophages [11-14].
In this variegate context, we asked whether non-mycobacterial TLR ligands could modify intracellular MTB viability in infected macrophages. This should be addressed to verify whether TLR ligands could be new target for TB drugs and to evaluate the risk of TLR ligand-mediated adjuvant use in MTB-infected individuals. In the present study we investigated the effects resulting from TLR2/6 engagement by a synthetic derivative of the Mycoplasma-derived macrophage-activating lipopeptide of 2 kDa (MALP-2) and from TLR4 triggering by lipopolysaccharide (LPS) on murine MTB-infected macrophages. MALP-2 derivatives are attractive candidate adjuvants for both mucosal and systemic vaccine formulations [15, 16]. In fact, MALP-2 derivatives induce secretion of pro-inflammatory cytokines/chemokines in DC [2, 17], enhance the capacity to process and present antigens to T and B cells [17], and increase memory adaptive immunity against co-administered antigens [15, 16]. Among MALP-2 derivatives, the S-[2,3-bispalmitoyloxy-(2R)-propyl]-R-cysteinyl-amido-monomethoxylpolyethyleneglyvcol, is a safe compound with low toxicity and pyrogenicity when administered in vivo [18]. The S-MALP-2 steroisomer is > 100 times less active than R-MALP-2 in stimulating the release of cytokines, chemokines and NO by macrophages [19]. Restricting TLR-induced signals is very important since a strong, protracted and non controlled stimulation of TLRs can induce excessive inflammation, which could damage host tissues and even be lethal, as for example in case of septic shock induced by LPS [18, 20]. Despite its high in vivo toxicity, LPS was used as a golden standard in our studies since it is one of the most potent inducer of macrophage activation in vitro. Moreover, LPS activates different pathways respect to those triggered by MTB TLR4 ligands [21] and activates NF-kB and IFN-responsive genes through MyD88-independent and -dependent pathways in macrophages [22].
MATERIALS AND METHODS
Microorganism
MTB H37Rv (ATCC 27294) was grown at 37°C in Middlebrook 7H9 medium under agitation (120 rpm) up to mid-exponential phase. Aliquoted stocks were stored at -70°C until use; the titers of stocks were verified on a regular basis by counting the numbers of CFU on Middlebrook 7H10 agar plates.
Reagents
LPS from E. coli was purchased from Sigma Aldrich Co, St Louis, MO. The MALP-2 derivative of 3,000 Da, S-[2,3-bispalmitoyloxy-(2R)-propyl]-R-cysteinyl-amido monomethoxylpolyethyleneglyvcol, was synthesized and purified by silica gel chromatography (>95%), as previously described [15]. This derivative was free from LPS contamination.
Preparation of Murine Bone Marrow-Derived Macrophage
Bone marrow-derived macrophages were prepared as previously described [23]. Briefly, bone marrow was flushed from femurs of healthy C57BL/6 mice with Dulbecco’s Modified Eagle’s Medium (DMEM) and cells were plated at 2 x 106 in 24-well plates in complete DMEM medium (cDMEM: Fetal Bovine Serum 10%, L-glutamine 1%, β-mercapto-ethanol) with the addition of 10% L-929-conditioned medium. After 1 day of incubation, the medium was replaced with antibiotic-free cDMEM supplemented with 10% L-929-conditioned medium. The cells were incubated for an additional 7 days, with medium replaced every 2 days before use in MTB-infection experiments.
Infection of Macrophages with MTB
Following the 7-day culture period, the macrophage cultures were infected with MTB, according to the following protocol: bacteria were diluted from frozen stocks in cDMEM and added at a multiplicity of infection (MOI) of 1:1 or 1:0.1 (macrophage to bacterium ratio) for 2 or 8 days of culture, respectively. MTB was co-incubated with macrophages at 37°C in 5% CO2 for 3 h and then washed 3 to 5 times in DMEM to eliminate extracellular bacteria. Following the last wash, cells were replaced in cDMEM and the stimuli (LPS and MALP-2) were added and left for the entire length of the experiments. Cells were infected with the two different MOI of infection in order to obtain after 2 or 8 days of culture a significant percentage of infected cells without evident sign of cell suffering (as checked by visualization of the macrophage culture by optical microscopy). After 2 or 8 days of incubation culture fluids were filtered through membranes (pore size 0.22 μm, Millipore, Malsheim, France) and then used for cytokine/chemokine and nitrite detection. The bacterial load was evaluated through colony forming unit (CFU) assays after 8 days of macrophage culture. This time point was selected to allow that MTB-triggered immune-evasion mechanisms, which reduce macrophage microbicidal power, could occur [14, 24].
CFU Assay
After 8 days of culture, the number of bacteria in macrophage culture was enumerated by CFU assay. Undiluted or diluted in distilled water culture supernatants were plated on Middlebrook 7H10 agar to evaluate the presence of external MTB. The intracellular MTB was revealed by lysing the macrophages with distilled water containing 0.1% saponin for 3 min, and then plating 10-fold dilutions in distilled water of the culture lysate on Middlebrook 7H10 agar. The MTB colonies were counted visually after 21 days of incubation. The value of CFU was the sum of both external and intracellular MTB CFU. In our macrophage cultures more than 96% of viable MTB were intramacrophagic, as calculated by enumerating MTB in supernatants and lysates.
Cytokine Detection
MTB-infected macrophage culture supernatants were harvested at 2 and 8 days and assayed for IL-6, IL-10, MIP-1-β, MIP-1-α and TNF-α by specific quantitative sandwich ELISA Kits (mouse Quantikine, R&D System, Abingdon, UK), in accordance with the manufacture’s instructions. Quantification was made against a standard curve obtained for individual cytokine standards provided by manufacturer.
Nitric Oxide Measurement
Culture supernatants were harvest at 48 h, and proteins were removed by ultrafiltration using 10,000 MW cut-off filters (Millipore). Nitric oxide concentrations were determined by colorimetric detection of nitrite as an azo dye product of the Griess Reaction using a specific colorimetric kit (ParameterTM Total NO/nitrite/nitrate, R&D System). To measure total nitrite, an enzymatic conversion of nitrate to nitrite by nitrate reductase was performed, in accordance with the manufacturer’s instructions.
RESULTS AND DISCUSSION
The effect of the TLR2/6 agonist MALP-2 derivative, tested at concentrations ranging from 5-500 ng/ml on the intracellular growth of MTB in bone marrow-derived murine macrophages. An optimal concentration of LPS (500 ng/ml) was also tested in parallel to gain strong macrophage activation through TLR4. We found that macrophage treatment with the lowest concentrations of the MALP-2 derivative (5-50 ng/ml) and LPS significantly reduced the intracellular growth of MTB as compared to untreated MTB-infected macrophages (P<0.01 by ANOVA-Bonferroni’s multiple t-test or Student’s t-test) (Fig. 1 A and B). However, the degree of microbicidal effect induced by lower MALP-2 derivative concentrations was significantly different from that conferred by LPS (P<0.05 LPS versus 50 ng/ml MALP-2 derivative and P<0.01 LPS versus 5 ng/ml MALP-2 derivative by ANOVA-Bonferroni’s multiple t-test). These data clearly demonstrated that the engagement of TLR2/6 and TLR4 with non-mycobacterial ligands can activate microbicidal mechanisms in macrophages, suggesting that TLR deserve to be investigated as new targets for anti-TB drugs. In addition, considering that TLR2/6 and TLR4 are targets for many bacterial compounds, our finding suggests that the capability of macrophages to kill intracellular MTB may be modulated by the presence of co-infection or the presence of commensal microbial flora in MTB-infected individuals.
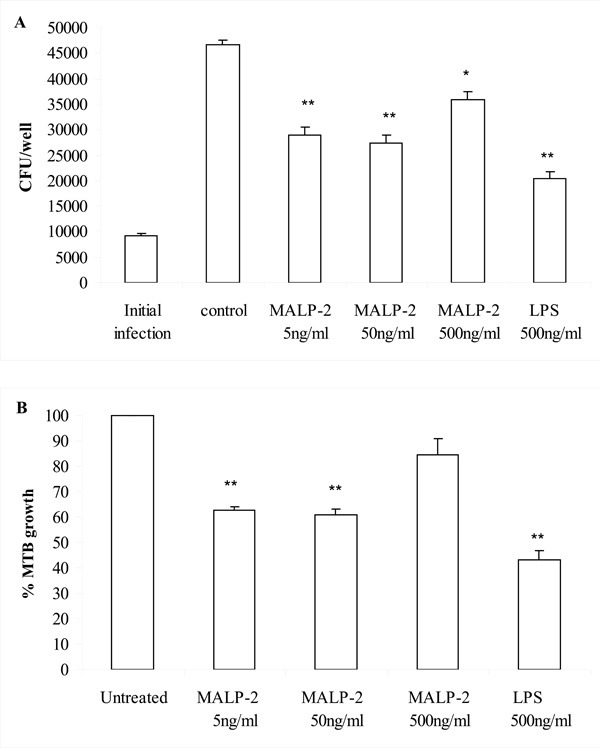
The MALP-2 derivative and LPS reduced intracellular replication of MTB in murine macrophages. Bone marrow-derived macrophages infected with MTB (MOI 1:0.1) were stimulated with either LPS or the MALP-2 derivative at the indicated concentrations. After 8 days, bacteria in the macrophage cultures were enumerated by CFU assay, as described in Materials and Methods. In panel A, the number of CFU is shown as mean ± SEM of triplicate determinations of a single representative experiment out of 4. In panel B, the percentages of MTB growth are shown as the mean ± SEM of 4 independent experiments. Responses that were significantly different from control untreated macrophages are indicated by asterisks as revealed by both ANOVA-Bonferroni’s multiple t-test and Student’s t-test (*, P<0.05; ** P<0.01).
Notably, the highest concentration of the MALP-2 derivative (500 ng/ml) resulted in a poor control of MTB replication (Fig. 1 A, B). An ANOVA-Bonferroni’s multiple t-test revealed a significant difference (P<0.01) of MTB growth inhibition induced by lower MALP-2 derivative concentrations compared to the effect caused by the highest concentration. In the search of a mechanism to explain why lower concentrations of MALP-2 derivative were more effective in reducing MTB intracellular growth, we measured the release of NO, the most powerful anti-mycobacterial defense mechanism in mice [25, 26]. In fact, a genetic disruption for the inducible NO synthase (iNOS) was associated with a significantly higher risk of dissemination and mortality in mice infected with MTB [27]. We found that the MALP-2 derivative, in a concentration dependent manner, and LPS, more potent, were NO inducers (Table 1). This suggests that the control mechanism for MTB intracellular growth may rely heavily upon NO, although this does not rule out the possibility of other contributing mechanisms. The microbicidal power of NO was however insufficient to induce MTB killing upon stimulation with the highest concentration of the MALP-2 derivative. In fact, despite a consistent NO production, no MTB load reduction was observed. In addition, the relatively weak NO induction observed after treatment with the lowest concentrations of MALP-2 derivative was sufficient to achieve intra-macrophagic killing of MTB, in comparison with the high level of NO induction observed after LPS treatment. Thus, a linear regression analysis revealed no correlation (R=0.2) between amount of NO produced and inhibition of MTB growth in macrophages treated with LPS or MALP-2 derivative. These data indicate that after reaching a critical threshold higher concentrations of MALP-2 derivative or LPS have no additional effect, rather they can even antagonize the NO-mediated killing of MTB. This is not completely surprising in view of the capacity of MTB to adapt to the NO synthesized by activated macrophages by either deploying NO scavengers or excluding iNOS from the phagosomes [28].
Cytokine, Chemokine and Nitrite in the Culture Supernatant Fluids of Murine MTB-Infected Bone-Marrow-Derived Macrophages Stimulated with the MALP-2 Derivative or LPS
| Stimuli | Days | MOI | TNF-α | IL-6 | IL-10 | MIP-1β | NO2 |
|---|---|---|---|---|---|---|---|
| pg/ml | pg/ml | pg/ml | pg/ml | µMol/L | |||
| Untreated | 2 | 1:1 | 0 | 188±62 | 92±2 | 1068±38 | 4.34±0.9 |
| 8 | 1:0.1 | 0 | 207±52 | 304±69 | 8078±148 | n.d. | |
| MALP-2 | 2 | 1:1 | 25±10 | 824±23* | 129±10 | 970±40 | 13.05±0.75* |
| 5 ng/ml | 8 | 1:0.1 | 0 | 211±53 | 260±15 | 7000±75* | n.d. |
| MALP-2 | 2 | 1:1 | 20±5 | 1243±7* | 102±6 | 1112±25 | 19.25±0.35** |
| 50 ng/ml | 8 | 1:0.1 | 0 | 301±44 | 418±90 | 6587±70* | n.d. |
| MALP-2 | 2 | 1:1 | 70±6* | 1470±64* | 126±6 | 1537+19* | 50.65±0.95** |
| 500 ng/ml | 8 | 1:0.1 | 10±5 | 538±151* | 361±89 | 5821±115* | n.d |
| LPS | 2 | 1:1 | 1879±39**,## | 2138±88**,# | 109±12 | 6846±225**,## | 142.4±1.8**,## |
| 500 ng/ml | 8 | 1:0.1 | 336±52**,## | 1284±69**,# | 504±91 | 6709±217* | n.d. |
a)Murine macrophages were infected with MTB at MOI 1:1 for 2 days or at MOI 1:0.1 for 8 days and cultured in the presence of LPS or the MALP-2 derivative at the indicated concentration. Culture supernatants harvested at the indicated time points were used for IL-6, IL-10, TNF-α , MIP-1β detection by specific quantitative sandwich ELISA Kits. The results are presented as mean.
± SEM in pg/ml. Pooled data of 3 independent experiments are shown. Nitrite was detected at 2 days with a specific colorimetric kit. The results are presented as mean ± SEM in µ Mol/L. Pooled data of 3 independent experiments are shown.
* p<0.05 and
** p<0.01, level of statistical significance between cytokine or nitrite release in stimulated versus untreated macrophages.
# p<0.05 and
## p<0.01, level of statistical significance between cytokine or nitrite release in LPS-treated versus MALP-2 derivative-stimulated macrophages.
On the other hand, the release of other mediators by activated macrophages may contribute to MTB survival independently from a direct effect on the NO pathways. Thus, we investigated macrophage production of IL-6, IL-10, TNF-α and MIP-1β. TNF-α is a cytokine critically important for host defense against MTB infection [29], which enhances release of reactive nitrogen intermediates from macrophages [30, 31]. However, the production of this cytokine does not always correlate with the extent of growth control in MTB-infected macrophages [32, 33]. Infection with virulent MTB strains induced the secretion of higher levels of bioactive TNF-α than attenuated strains, which in turn correlates with their ability to multiply intracellularly. Treatment of human infected macrophages with anti-TNF-α antibodies reduces the growth rate of intracellular bacteria, whereas bacterial replication is augmented by addition of exogenous TNF-α [34]. On the other hand, MIP-1β was found to suppress intracellular MTB growth and its diminution may promote intracellular survival of MTB in human macrophages [35]. IL-6, a cytokine required for protection against MTB infection [36], can reduce the activation of uninfected macrophages, thereby contributing to the inability of the immune response to eradicate infection [37]. Finally, IL-10, an important negative regulator of Th1 immunity in tuberculosis [38], can also inhibit macrophage activation [39]. With this background, we measured the production of the above cytokine by MTB-infected cells, treated or untreated with the MALP-2 derivative or LPS. We found that after 2 days of culture, LPS greatly induced production of IL-6, MIP-1β and TNF-α in MTB-infected macrophages (Table 1). Although to a lesser extent than that observed for LPS, the highest concentrations of the MALP-2 derivative enhanced release of these three cytokines, whereas only IL-6 secretion was enhanced at low MALP-2 derivative concentrations (Table 1). After 8 days of culture, we observed that upon LPS stimulation, the accumulation of both IL-6 and TNF-α was still greater compared to untreated MTB-infected macrophages, whereas MIP-1β accumulation was reduced (Table 1). Similar results, with the exception of almost undetectable TNF-α secretion, were obtained at the highest concentrations of the MALP-2 derivative, whereas the lower concentrations of MALP-2 reduced only, in a concentration dependent manner, the accumulation of MIP-1β. The accumulation of IL-10, after 2 or 8 days of culture, was modified neither by LPS nor by the MALP-2 derivative.
Although the secretion of these mediators does not allow a clear elucidation of their correlation with MTB viability, we observed that, the enhancement of MTB intra-macrophagic killing by the MALP-2 derivative is unrelated to the production of these selected cytokines. In fact, the MALP-2 derivative killed intracellular MTB without the need to secrete TNF-α and by inducing only very weak IL-6 secretion. This is an important advantage in the context of potential in vivo application, since TNF-α is the main responsible for LPS-mediated lethal toxicity [20] and IL-6 is a potent pyrogenic mediator [40]. Moreover, we can speculate that the enhancement of IL-6 and TNF-α secretion observed in MTB-infected macrophages upon stimulation with either LPS or the highest concentration of MALP-2 may have promoted mechanisms favoring MTB replication at the expense of NO-mediated killing.
In conclusion, the MALP-2 derivative, at low concentrations, reduces intracellular growth of MTB without the need of releasing pro-inflammatory mediators implicated in systemic toxicity. Our data also suggest that it can be proposed the safe use of the MALP-2 derivative as adjuvant in MTB-infected people, since it has not increased MTB replication, even at the highest concentrations tested. The opportunity to modulate macrophage anti-mycobacterial defense mechanisms through low toxic non-mycobacterial TLR ligands opens favorable perspectives towards the development of new anti-MTB drugs, which are urgently needed for this dreadful disease.
ACKNOWLEDGEMENTS
This study was supported in part by the grant H51 from the Ministero della Salute (Italy) and the grant LSHP-CT-2003-503240 (MUVAPRED Project, European Community).
We are grateful to Giovanni Piccaro and Antonio Di Virgilio for mouse housing and technical assistance.

