All published articles of this journal are available on ScienceDirect.

Influence of Fecal Sample Storage on Bacterial Community Diversity
Authors Info & Affiliations
Abstract
Previous studies have identified a correlation, either positive or negative, between specific stool bacteria strains and certain autoimmune diseases. These conflicting data may relate to sample collection. The aim of this work was to evaluate the influence of the collection parameters of time and temperature on bacterial community composition. Samples were taken from healthy children and immediately divided in 5 sub-samples. One sample was frozen immediately at -80°C, while the other aliquots were frozen 12, 24, 48, and 72h later DNA extracted from each sample was used to amplify the 16S rRNA with barcoded primers. The amplified products were pooled and partial 16S rRNA sequences were obtained by pyrosequencing. Person-to-person variability in community diversity was high. A list of those taxa that comprise at least 1% of the community was made for each individual. None of these were present in high numbers in all individuals. The Bacteroides were present in the highest abundance in three of four subjects. A total of 23,701 16S rRNA sequences were obtained with an average of 1,185 reads per sample with an average length of 200 bases. Although pyrosequencing of amplified 16S rRNA identified changes in community composition over time (~10%), little diversity change was observed at 12 hours (3.06%) with gradual changes occurring after 24 (8.61%), 48 (9.72%), and 72 h (10.14%), post collection.
INTRODUCTION
Associations between gut microbiota and either the risk for or presence of specific human diseases have been shown in recent years. Several allergenic and autoimmune diseases including Crohn's disease [1, 2], celiac disease [3], and type 1 diabetes [4] have shown correlations with an altered gut microflora in humans and animal models of these disorders. Bacteria inhabiting the gastrointestinal tract may affect mucosal permeability and/or lead to abnormal immunoregulation as has been reported in multiple sclerosis, allergy and inflammatory bowel disease [5]. The influence of gut bacteria on human disease is not limited to immunologically mediated disorders as altered gut microbiota have also been related to a propensity for obesity [6].
The microbial composition of the gastrointestinal tract has been studied by a variety of molecular or culture based techniques using either fecal samples or samples obtained by biopsy. Biopsies are invasive. On the other hand, fecal samples are easier to obtain and contain intestinal bacteria and exfoliated epithelial cells that may provide useful information concerning the gastrointestinal tract. However, the analysis of the microbiota in fecal samples requires that the sampling procedure does not alter the microbial composition of the samples.
In order to evaluate whether microbial communities maintain the same structure after storage at room temperature, stool samples from healthy individuals were collected with aliquots stored at room temperature at various times up to 72 hours. By using 16S rRNA PCR amplification and pyrosequencing, changes in community composition were analyzed.
MATERIALS AND METHODS
Sample Collection, Experimental Design, and DNA Extraction
Fresh fecal samples were obtained from four healthy children at the University of Turku Hospital (Turku, Finland). Samples were collected in sterile flask containers and immediately divided in 5 sub-samples. One sample was frozen immediately at -80°C and the other aliquots were kept at room temperature and frozen after 12, 24, 48, and 72h. The samples were then sent on frozen ice packs to the University of Florida for further analyses. Bacterial DNA was isolated from the stool samples using the FastDNA® Kit (Qbiogene, Inc., CA). Following DNA extraction, samples were purified with DNeasy Tissue kits (Qiagen, Valencia, CA) utilizing the manufacturer’s instructions.
16S rRNA Gene Amplification and 454 Pyrosequencing
The V3 region of the 16S rRNA gene was amplified from the stool DNA. To obtain 16S rRNA sequences from many samples with a single 454 pyrosequencing run, an 8-base barcode was added to the 5’-end of the reverse primers using the self-correcting barcoding method of Hamady et al. [7]. The barcoded 16S rRNA primers were attached to the 454 Life Sciences® primer B and A, and a two-base linker sequence was inserted between the 454 adapter and the 16S rRNA primers to reduce any effect the composite primer might have on PCR efficiency (see supplementary material). Six independent PCR reactions were performed for each sample. The PCR conditions were 94°C for 2 minutes, 25 cycles of 94°C for 45s; 55°C for 45s; and 72°C for 1 min extension; followed by 72°C for 6 minutes. The six PCR replications for each of the 20 samples were combined and purified with the QIAquick PCR Purification Kit (Qiagen, Valencia, CA). The DNA concentration of the PCR products was measured using on-chip gel electrophoresis with an Agilent 2100 Bioanalyzer and DNA LabChip® Kit 7500. Finally the reactions were combined in equimolar ratios to create a DNA pool that was used for pyrosequencing from adaptor A.
Data Preparation Prior to Further Analysis
Initially, all pyrosequencing reads were screened for quality and length of the sequences. The ends of the sequences with Phred scores lower than 20 were trimmed and reads shorter than 100 bases were removed from the dataset using Trim2 [8]. The trimmed sequences were than screened beginning with the 8-base barcode. A custom perl script was written to find the barcode and generate a new file for each sample. The sequences were then relabeled accordingly to denote the original sample.
For each of the 20 libraries, the sequences were grouped into Operational Taxonomy Units (OTUs) using the same pipeline applied by Roesch et al. [9] and by defining an OTU as those sequences with a similarity equal to, or greater than, 97%. The longest sequence in a cluster was chosen as a representative sequence for each cluster and a new non-redundant library was created for each sample. The number of OTUs in each library was determined in each sample for further analysis.
UniFrac Significance Test and Phylogenetic P-Test
UniFrac calculations were performed separately on the samples from each individual (subjects A, B, C and D). The analysis was done in a pairwise hierarchical scheme that compared each of the time points from 12-72 h against the immediately frozen sample as a reference. For each pairwise comparison, MUSCLE [10] was used to build a guide tree using the UPGMA agglomerative clustering method. The resulting phylogenetic tree from MUSCLE, a text file containing the number of sequences found within each out, and the subject from which the samples were derived were uploaded into the online UniFrac program [11]. The weighted and unweighted UniFrac significance tests and a phylogenetic P-test were performed. The P-values reported for multiple comparisons and were corrected by Bonferroni correction, which is performed by multiplying the raw P-value by the number of permutations.
Analysis of Molecular Variance (AMOVA)
To examine variation in the genetic structure of the bacterial population during time of storage at room temperature, AMOVA analysis was performed using the same dataset that was imported into UniFrac. A list of aligned representative sequences and their frequencies within each sample were placed in a text file that was used to perform the analysis of molecular variance. The AMOVA procedure incorporates the estimated divergence between sequences and their frequencies. All calculations were performed using Arlequin ver. 3.0 [12]. With AMOVA, the variance between communities and the percentage of total variance between communities was determined.
Fraction of OTUs Shared
The proportion of OTUs shared among the communities was determined using SONS [13], which uses the .list output files from DOTUR as input and determines the fraction of OTUs shared by communities as a function of genetic distance. For this analysis, all sequences were pooled from each subject in the same file and aligned using MUSCLE [10]. A distance matrix was then generated using the PHYLIP version 3.6 [14]. With the distance matrix, DOTUR [15] was used and the output files from DOTUR were used to run SONS. A list of shared OTUs comprising the number of sequences from each OTU at different time of storage was then generated. To identify changes in specific bacterial lineages and obtain a quantitative description of the bacterial lineages, only those OTUs that were 1% or more abundant were used in the dataset. For each OTU (defined as 97% similarity or greater) an exact Chi-square test (based on 50000 Monte Carlo iterations) was calculated to obtain a p-value for the null hypothesis. That is, H0: equal success probabilities, which means that there are no differences in that specific OTU after sample storage at room temperature. The p-values were ordered and processed to find a false discover rate (FDR) less than 1%.
Phylogenetic Classification
The 16S rRNA gene sequences were phylogenetically assigned according to their best matches to sequences in the NAST (Nearest Alignment Space Termination) database [16]. The sequences were submitted to a web-interface tool (http://greengenes.lbl.gov/NAST) and aligned against the greengenes 16S rRNA reference database (188,073 aligned 16S rRNA records). Once aligned, the sequences were taxonomically classified according to the best match with the reference database using the NCBI taxonomic nomenclature.
RESULTS
The pyrosequences averaged 200 bases in length. The number of sequences and the number of OTUs found (97% similarity) for each sample was determined (Table 1). After removing short reads, trimming low quality ends, and removing chimeric sequences, a total of 23,701 reads were available for the analyses. The average number of reads for each sample was 1,185 and the number of OTUs found varied form a maximum of 198 (subject A at 12h) to a minimum of 14 (subject D 1h).
Number of Sequences Sampled and Number of OTUs Found at 97% Similarity
| Subject | No. of Sequences Sampled | No. of OTUs Found |
|---|---|---|
| A-IM | 1674 | 160 |
| A-12h | 1891 | 198 |
| A-24h | 1261 | 146 |
| A-48h | 1285 | 137 |
| A-72h | 1290 | 145 |
| B-IM | 1291 | 193 |
| B-12h | 1345 | 157 |
| B-24h | 1030 | 92 |
| B-48h | 1155 | 64 |
| B-72h | 1649 | 117 |
| C-IM | 1558 | 131 |
| C-12h | 849 | 74 |
| C-24h | 826 | 89 |
| C-48h | 1379 | 98 |
| C-72h | 1031 | 66 |
| D-IM | 1113 | 14 |
| D-12h | 1017 | 20 |
| D-24h | 730 | 18 |
| D-48h | 638 | 23 |
| D-72h | 689 | 19 |
P-values Calculated by the UniFrac Significance Test from Comparisons of the Bacterial Community Found in Stool Samples Immediately (IM), after 12h, 24h 48h and 72h Prior to DNA Extraction and Amplification of the16S rRNA
| Subject A | Subject B | Subject C | Subject D | |||||
|---|---|---|---|---|---|---|---|---|
| Qualitative | Quantitative | Qualitative | Quantitative | Qualitative | Quantitative | Qualitative | Quantitative | |
| p-values | ||||||||
| IM x 12h | 1.00 | 1.00 | 1.00 | 0.74 | 1.00 | 0.98 | 0.93 | 1.00 |
| IM x 24h | 1.00 | 1.00 | 0.95 | 0.26 | 1.00 | 0.95 | 0.81 | 0.92 |
| IM x 48h | 0.59 | 0.93 | 0.32 | 0.09 | 0.73 | 0.97 | 0.23 | 0.63 |
| IM x 72h | 0.94 | 0.98 | 1.00 | 0.04 | 0.98 | 0.98 | 0.35 | 0.82 |
The comparison was made considering the presence/absence (qualitative) and the abundance (quantitative) of taxonomic unities. All P-values have been corrected for multiple comparisons by multiplying the calculated P-value by the number of comparisons made (Bonferroni correction). For each pairwise comparison, if the p-value is less than or equal to 0.001 the result indicates a highly significant difference in the composition of the communities sampled by each library. P-values from 0.001 to 0.01 indicate a significant difference, from 0.01 to 0.05 indicate a marginally significant difference, from 0.05 to 0.1 indicate a suggestive difference and if the p-values are greater than 0.1 the result indicate that the difference is not significant.
P-values Calculated by the P-Test from Comparisons of the Bacterial Community Found in Stool Samples Immediately (IM), after 12h, 24h 48h and 72h prior to DNA Extraction and Amplification of the16S rRNA
| Subject A | Subject B | Subject C | Subject D | |
|---|---|---|---|---|
| p-values | ||||
| IM x 12h | 0.79 | 0.23 | 0.99 | 0.99 |
| IM x 24h | 0.99 | 0.01 | 0.90 | 0.72 |
| IM x 48h | 0.10 | ≤ 0.01 | 0.09 | 0.33 |
| IM x 72h | 0.19 | ≤ 0.01 | 0.08 | 0.59 |
P-values have been corrected for multiple comparisons using the Bonferroni correction. (Each p-value was multiplied by the number of pairwise comparisons performed). For each pairwise comparison, if the p-value is less than or equal to 0.001 the result indicates a highly significant difference in the composition of the communities sampled by each library. P-values from 0.001 to 0.01 indicate a significant difference, from 0.01 to 0.05 indicate a marginally significant difference, from 0.05 to 0.1 indicate a suggestive difference and if the p-values are greater than 0.1 the result indicate that the difference is not significant.
AMOVA Statistics Showing Variance Between Groups and Percent of Total Variance for Each Pairwise Comparison within the Four Subjects Tested
| σ2b | % of σ2T | |
|---|---|---|
| Subject A | ||
| IM x 12h | 0.98 | 2.00 |
| IM x 24h | 0.92 | 1.98 |
| IM x 48h | 2.84 | 5.35 |
| IM x 72h | 1.97 | 3.85 |
| Subject B | ||
| IM x 12h | 3.36 | 5.88 |
| IM x 24h | 11.00 | 18.36 |
| IM x 48h | 11.00 | 18.62 |
| IM x 72h | 8.87 | 14.42 |
| Subject C | ||
| IM x 12h | 2.20 | 3.67 |
| IM x 24h | 3.41 | 5.58 |
| IM x 48h | 2.35 | 4.02 |
| IM x 72h | 4.13 | 6.90 |
| Subject D | ||
| IM x 12h | 0.21 | 0.70 |
| IM x 24h | 2.78 | 8.05 |
| IM x 48h | 4.14 | 10.88 |
| IM x 72h | 5.87 | 15.40 |
All P values obtained were < 0.001. Levels of significance were based on 10,000 iteration steps; σ2b = variance between groups; σ2T = total variance.
Overall Bacterial Community Comparisons
The UniFrac p-values were determined for both presence/absence and abundance of bacterial lineages (Table 2). The UniFrac significance test accounts for both the tree topology and the branch lengths, and tests the hypothesis that there has been more unique evolution within each environment than would be expected if the sequences were randomly distributed among the environments [11]. If the p-values are greater than or equal to 0.1, the communities are not considered statistically significantly different. From subjects A, C and D, the p-values were greater than 0.1 at all time periods in both qualitative and quantitative analysis. Thus, no changes in bacterial community were observed even after 72h. In subject B, no qualitative changes were observed after 72h. However, a significant quantitative difference was observed when comparing the IM to 48h time points.
UniFrac compares communities for significant differences and typically does not detect shifts in bacterial communities when there are many sequences that are closely related to each other but that are unique to one particular sample. Thus, the phylogenetic P-test is more likely to give significant results when fewer OTUs are required to explain the actual distribution of sequences in environments than would be required if sequences were randomly assigned to samples [11]. The p-values calculated for each pair of samples were determined using the Phylogenetics P-test (Table 3). No significant changes were observed in bacterial community compositions in any of the subjects after 12h of sample storage (Table 3). A significant difference between the original bacterial community and the community observed after 24h in subject B was observed (p-value = 0.01). However no significant differences were found in the other subjects at 24h. Most significant differences were found after 48h. The subject B samples changed significantly beginning at 24h while subject D’s samples showed no changes over the 72h period. The samples from subjects A and C showed significant changes at 48h.
Genetic Differentiation of Bacterial Communities
Changes in bacterial communities were also analyzed at the level of community genetic structure inferred by analysis of variance. The Analysis of Molecular Variance (AMOVA) computes differences between microbial communities based on phylogenetic information. However, unlike UniFrac, which calculate the population differentiation based on tree topology and branch lengths, AMOVA makes use of the molecular information obtained in the DNA sequences to investigate the genetic differentiation of the sampled communities. In addition to determining whether a change has occurred in bacterial community composition, AMOVA describes the extent to which the entire community has changed.
After 12, 24, 48, and 72 h, the average bacterial community across all subjects changed by 3.06, 8.61, 9.72, and 10.14%, respectively (Fig. 1, Table 4). All of these changes were statistically significant. Most of the change in community structure occurred between 12 and 24 h after collection.
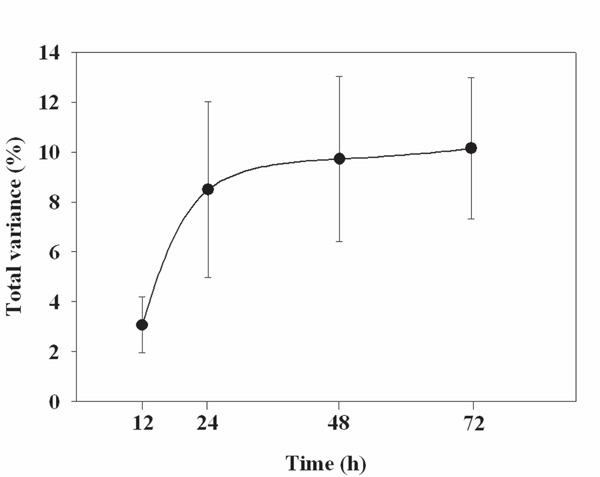
The % change in bacterial community composition in the stool samples compared to samples frozen immediately. Each point is the mean of samples from four individuals with the bars representing the standard error about the mean.

Heat map showing the proportion of bacteria found at different times of storage (immediately (IM), after 12h, 24h, 48h and 72h) and the significance of the variation observed. Maps A, B, C and D represent the subjects analyzed. The scale on the right represents the contribution of a particular OTU and it is expressed in percentage of the total. The closest bacterial relative is shown in the left side of the map. Note that OTUs were classified to the highest specificity level, ranging from phylum to genus.
Identification of the Bacterial Taxa whose Abundances Change Over Time
In order to identify which bacterial lineages contribute to differences observed after storage 16S rRNA sequences were compared and clustered based on 97% similarity level and above. For each comparison, an exact Chi-square test was performed to obtain p-values which were used to determine whether the relative abundance of specific groups of bacteria changed over time. Finally, representative sequences for each OTU were classified and closest bacterial relative identified. Taxonomic units were classified to the highest specificity level, ranging from phylum to genus. A heat map showing the proportion of bacteria found at different times of storage and the significance of the variation was prepared (Fig. 2). To avoid errors in analysis based on undersampling of certain taxa, only those taxa that represented at least 1% of the total number of sequences in any subject at any time point were considered.
Those taxa that represent more than 1% of the total population were identified to the genus level wherever possible (Fig. 2). In some cases, taxa could only be identified at the family or order level. The proportion of identifiable bacteria that also represent more that 1% of the population varied among subjects and across time with as few as 63.2% in Subject A at 48h or 94.2% in Subject D at 24 h.
Although most of the 454 sequences could be identified at the genus level, most could not be classified at the species level. However, genus-level identification allows us to estimate the general physiological characteristics of these organisms. No genus was represented by more than 1% of sequences in all four subjects. This is largely owing to the lack of diversity in the subject D samples. Only five taxa representing over 90% of the bacterial communities across all time points could be identified in the subject D samples.
Some taxa clearly change across time in more than one subject. For example, Rumminococcus strains decline in subjects B and C over time. Alistipes and Faecalibacterium strains decline over time in subjects A and B. The bacterial changes in these samples across time are not always consistent between samples. For example, in subject A, the Dialister and Streptococcus organisms increased in relative abundance over time, while in sample B, the abundance of these organisms dropped rapidly over time.
Differences between subjects were greater than the variation in the fecal sample variation caused by storage within the same subject. Irrespective of the subject analyzed, most of the organisms were members of Firmicutes and Bacteroidetes phyla. Most Firmicutes were members of Clostridium. The variability in the relative abundance of bacteria found within individuals increased or decreased over time depending of the bacterial phylotype. The most abundant genera identified, Bacteroides and Clostridium, decreased over time while certain species from the Enterobacteriaceae increased.
DISCUSSION
In recent years, there has been increasing interest in the discovery of correlations between bacterial communities in the human gut and disease [2, 6, 17-23]. Several studies have shown a correlation between gut bacteria communities and the development of autoimmune diseases. In Crohn’s disease, a T-RFLP fingerprinting method was used to shown that bacterial diversity is higher in stool from healthy subjects compared to their twin diseased subjects. Two rodent models for type 1 diabetes have shown that feeding antibiotic or probiotics can delay the onset of the disease [1, 24-27]. In addition, a germ-free environment increases the incidence of diabetes in NOD mice [28, 29].
These recent results encourage the use of human stool samples to search for bacteria that are correlated, negatively or positively, with the onset of type 1 diabetes in children. Two studies have collected such samples that could be used for this purpose. The Diabetes Prediction and Prevention study (DIPP) in Finland has been collecting stool and blood samples quarterly (beginning soon after birth) from children at high risk for the disease since 1994. A larger prospective study in the United States, Germany, Sweden, and Finland called The Environmental Determinants of Diabetes in the Young (TEDDY) (seeking to determine possible environmental triggers of autoimminty) has been collecting samples since 2004 from genetically high risk children on a monthly basis [30]. In both studies, some have seroconverted and developed type 1 diabetes.
The stool samples from the DIPP and TEDDY studies could be very useful in determining whether any bacteria are correlated with both the triggering of the autoimmune process and/or the onset of type 1 diabetes. However, to be useful, the conditions under which they were collected need to be examined to be certain that collection protocols to not impact the bacterial contents of these samples.
Four samples were taken from healthy children enrolled in the DIPP study. Each sample was divided into five aliquots. One was immediately frozen at -80°C while the others were frozen after 12, 24, 48, and 72h. The 72h period is the maximum time required to collect a sample from the infant or child until it is frozen at a DIPP or TEDDY repository. During that period of transit, the sample sits at or near room temperature. In order for DIPP or TEDDY samples to be useful, it is important to know whether the bacterial community composition changes over the 72-hour period.
Over a 72-hours, we showed that bacterial communities only change by ~10% with most change beginning between 12 and 24 hours. As expected, there was enormous variability from person to person in both bacterial community composition and in the rate of change in composition over time. This suggests that nutrient availability in stool is very different from sample to sample, thereby affecting growth rates at room temperature over time.
In addition, changes are observed only in the most abundant taxa. The samples are so diverse that no statistical changes are observed over 72hr if the whole community is considered. However, careful examination of the dominant taxa shows that changes are occurring.
These data suggest that new experiments may be needed to examine these issues further. For example, what is the effect of storage temperature over time? That is, could a delivery time of 2-3 days from the time of collection to the time of permanent storage be tolerated if the samples were immediately placed on ice packs? Is storage at 4°C or -20°C during transit cold enough to minimize changes in community composition?
Finally and most importantly, are the changes observed here over time, large enough to mask changes that may occur over time in one person during the several month period of seroconversion? TEDDY metadata shows that 23.9% of all samples collected in the Georgia, Florida, Colorado, and Washington State clinics are frozen within 24h of collection. Thus, if AMOVA shows that the changes that occur in bacterial community composition during seroconversion are much greater than 10%, the current DIPP and TEDDY samples may be very useful, particularly the large number of samples frozen within 24h.
ACKNOWLEDGEMENTS
This work was funded by the National Institutes of Health (RO3 A142288), the Brehm Coalition for Type 1 Diabetes Research, and the Florida Agricultural Experiment Station.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.

