All published articles of this journal are available on ScienceDirect.

AFM Specific Identification of Bacterial Cell Fragments on Biofunctional Surfaces
Authors Info & Affiliations
Abstract
Biointerfaces with a highly sensitive surface designed for specific interaction with biomolecules are essential approaches for providing advanced biochemical and biosensor assays. For the first time, we have introduced a simple AFM-based recognition system capable of visualizing specific bacterial nanofragments and identifying the corresponding bacterial type. For this we developed AFM-adjusted procedures for preparing IgG-based surfaces and subsequently exposing them to antigens. The AFM images reveal the specific binding of Escherichia coli cell fragments to the prepared biofunctional surfaces. Moreover, the binding of bacterial cell fragments to the affinity surfaces can be characterized quantitatively, indicating a 30-fold to 80-fold increase in the quantity of bound antigenic material in the case of a specific antigen-antibody pair. Our results demonstrate significant opportunities for developing reliable sensing procedures for detecting pathogenic bacteria, and the cell can still be identified after it is completely destroyed.
1. INTRODUCTION
Because molecular recognition between specific receptors and ligands plays a key role in biological processes, developing bioselective surfaces based on this recognition is a large challenge in science and applications [1, 2]. Atomic force microscopy (AFM) has opened a wide range of novel possibilities for exploring biofunctional surfaces and biomolecules [3, 4] and is currently widely used to solve fundamental and applied problems in the biological sciences. Because AFM is capable of molecular-resolution imaging of single particles binding to a surface, it can be used as a research tool for investigating processes that occur on the surfaces of biosensing devices. Moreover, AFM itself can be used to detect bacterial antigens with the high accuracy.
Detecting pathogenic bacteria remains one of the challenging tasks in the applied biological and medical sciences [1, 5, 6]. The basic principles of developing biofunctional surfaces responsible for the specific binding with a microbiological analyte in model systems for the purposes of biological detection have been studied using different techniques for investigating surfaces including quartz crystal microbalance, surface plasmon resonance, and ellipsometry [5, 7-13]. These techniques are based on immobilizing specific antibodies on a substrate surface and subsequently exposing it to an antigen-containing suspension.
The proper orientation and immobilization of antibodies determines the sensitivity and specificity of the recognition system for the analytes of interest [14]. There are three main strategies for immobilizing antibodies on a solid support: direct adsorption, the use of avidin-biotin binding, or a self-assembled monolayer [7]. Direct adsorption, the simplest technique for depositing antibodies, yields the lowest affinity of the resulting layer because of the chaotic positioning of the antigen-binding region (Fab). But immobilizing protein G beneath the antibodies significantly improves their organization into a monolayer because of the ability of protein G to specifically bind IgG in the Fc region [15]. As a result, the Fab regions of immunoglobulin molecules in a layer are exposed outside the surface of the substrate, significantly increasing the resulting functionality of the layer. This approach has been widely used to investigate the specific binding of bacterial cells using QCM, SPR, ellipsometry, and other surface-based techniques [16], where a significant change in the signal (whose nature depends on the technique used) indicates the binding of bacterial cells to the surface.
Direct high-resolution studies of the process of the specific binding of a microbiological analyte to a biofunctional surface are still lacking. One of the most appropriate exceptions is the AFM and SEM investigation of Salmonella typhimurium specific adhesion to an anti-Salmonella antibody-modified surface [17]. These results together with multiple works on the AFM of microbial surfaces (e.g., [18-20]) show that AFM is a powerful and effective tool for directly studying the functioning of biosensor surfaces.
In our work, we used AFM to investigate antibody-based biofunctional surfaces before and after their exposure to antigen-containing suspensions. To the best of our knowledge, whole bacterial cells were used as antigens in previous works on surface-based microbiological detection [12, 13, 21-24]. Our goal here was to study the specific binding of small bacterial fragments, which is a relevant task for microbiological biosensing for two reasons: first, it will allow detecting the specified pathogen based on part of it (or on a destroyed cell); second, it will allow intentionally destroying the cells before they are analyzed. We have therefore also modified the mode of bacterial cell fragmentation for further AFM imaging.
2. MATERIALS AND METHODS
2.1. Obtaining Bacterial Fragments
In this work, we used Escherichia coli 08 and Bacillus subtilis cells from the collection of the State Research Center of Applied Microbiology and Biotechnology. Microbial cells were grown for 16-20 hours on LB broth (ICN Company) at a temperature of 37ºC, precipitated by ultracentrifugation at 9300 g for five minutes (for Escherichia coli) or ten minutes (for Bacillus subtilis), and washed three times with physiological solution in the same mode. The obtained precipitate was resuspended in distilled water until the concentration of cells reached 109 cells/ml according to the opacity standard.
Cells were disrupted and their fragments were obtained in two ways. The ultrasound method involved exposing the suspension in the disintegrator Virsonic-100 (Virtis Company) in an ice bath with alternating 30 seconds of ultrasound and a 30-second pause for five minutes.
The other way involved the freeze-thaw method. The cell suspension in water (1 ml) was frozen at a temperature of -80ºC for one hour and then warmed in a water bath at room temperature. This procedure was repeated five times. Undestroyed cells were separated by centrifugation at 5000 g. Disrupted cells were inspected using a light microscope with immersion and a 90-fold zoom of the objective.
2.2. Preparing the Biofunctional Surface
Freshly cleaved mica was first modified in a glow discharge (current 0.2 mA, V=1.5 kV) for one minute and then exposed to a droplet (20 μl) of protein G solution (concentration 0.6 mg/ml) in Tris-HCl buffer (20 mM Tris, 200 mM NaCl, pH 8.0) for one minute. The protein G modified mica plate was washed for one minute in a droplet (50 μl) of distilled water and then exposed to an anti-Escherichia coli monoclonal antibody (the clone number is C1H3) solution (concentration 0.1 mg/ml) in the Tris-HCl buffer (pH 8.0) for ten minutes. This surface was washed one more time in a distilled water droplet (50 μl) for one minute and then dried in a nitrogen flow.
For production of monoclonal antibodies IgG was used as described before [25]. This procedure included additional purification by the protein A/G that favors the formation of more stable antibody layer on protein G.
2.3. Exposing the Biofunctional Surfaces to the Analyte
We used three types of analyte suspensions in our experiments: suspensions containing Escherichia coli or Bacillus subtilis bacterial fragments in buffer and the buffer solution (pH 8.0) without antigens. The obtained biofunctional surfaces were exposed to a droplet (20 μl) of analyte suspension for ten minutes at a temperature of 32ºC. These pH and temperature values favor the specific binding reaction [26]. The mica plate was then immersed in the buffer solution and rotated in the shaker at 100 revolutions per minute for ten minutes. The mica surface was finally rinsed in a droplet (100 μl) of distilled water to avoid possible salt deposition upon drying of the buffer and then dried in a nitrogen flow.
2.4. AFM Imaging
All experiments were performed using a Nanoscope III multimode atomic force microscope (Veeco Instruments Inc., USA) in contact mode in air. Commercial silicon CSC11 (spring constant 0.35 N/m) cantilevers (MikroMasch) were used. The FemtoScan software (Advanced Technologies Center, Russia) was used for image processing. The scan rate was typically 2 Hz with 512 lines.
3. RESULTS AND DISCUSSION
To obtain surfaces that are affinitive to bacterial cells, we used a conventional approach based on depositing monoclonal antibodies on top of a protein G modified substrate [16]. Because protein G can specifically bind the heavy chain of immunoglobulin G in the Fc region [15], it fosters IgG molecules to expose their antigen-binding sites on the surface; the presence of protein G thus increases the total functionality of the IgG layer. But in this work, we introduced some additional stages of affinity surface preparation and adjusted the protein G and IgG deposition parameters to increase the resulting surface specificity and sensitivity to an antigen. The developed procedure is described in the Materials and Methods section. We gave much attention to arranging the conditions for modifying the mica in the glow discharge, depositing the antibody layer, and rinsing the surface in different specimen preparation stages (the rinsing procedure and duration).
We used muscovite mica as a substrate in our experiments. The mica surface has a natural negative charge in an aqueous environment. The protein G molecule at pH 8.0 also has a total negative charge (the pI of protein G is 5.0). We did not use coagulants and crosslinking agents to fix protein G on the mica surface. Those fixatives cause substantial conformational changes of protein [27]. We therefore treated freshly cleaved mica in a glow discharge before depositing the protein G layer (see Materials and Methods). This makes the adsorption of protein more effective because different chemical bonds on the mica surface are activated [28-30]. The antibody layer was deposited at pH 8.0, which is the optimal value for the specific binding of protein G with the Fc region of IgG [31]. The pH value 8.0 is very important because lowering the pH causes a reversal of this binding.
Topography of all successive steps of affinity surface preparation (including glow-discharged mica, protein G layer and IgG layer) and the results of scratching experiments on the final surface are presented in Fig. (1). Anti-Escherichia coli monoclonal antibodies were used in all the presented experiments. The resulting layer remained stable in an aqueous environment and after the rinsing procedure. The total thickness of the final layer is slightly more than 6 nm (and 1.5 about nm for the protein G layer alone), and its surface root mean square roughness is about 0.6 nm. The observed height agrees well with the height of the similar structure consisting of protein A and anti-Salmonella antibody reported by Lee et al. [17].
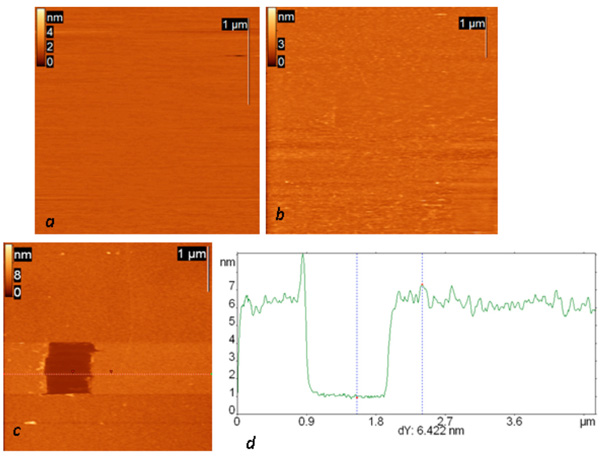
AFM height images of a mica surface modified in glow a discharge (current 0.2 mA, duration one minute) (a) and successively exposed to a protein G solution (concentration 0.1 mg/ml) (b) and then (after rinsing) to a solution of anti-Escherichia coli monoclonal antibodies (c). The dark square in (c) is a result of scratching experiments on this surface; the height difference is 24 nm. (d) Cross section along the dotted line in the AFM image.
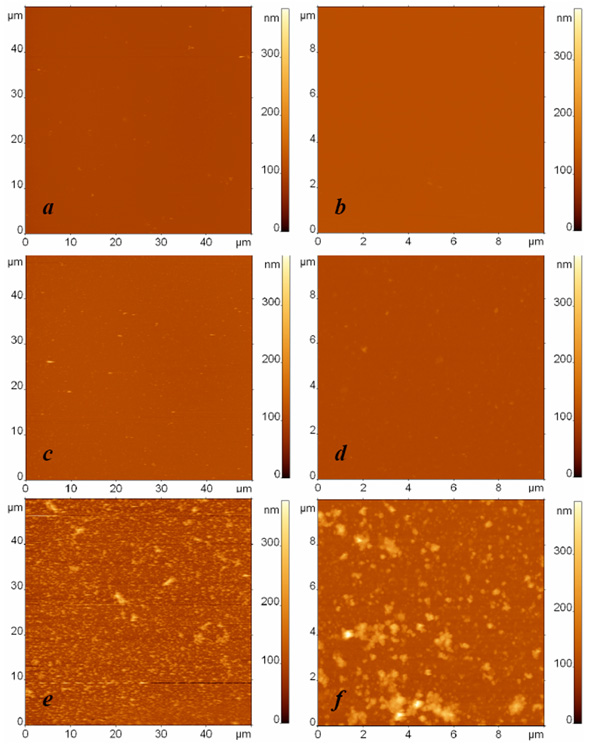
AFM images of surfaces modified with protein G and anti-Escherichia coli antibodies and exposed to (a, b) the buffer, (c, d) the suspension containing Bacillus subtilis cell fragments with the concentration 108 cells/ml, and (e, f) the suspension containing Escherichia coli cell fragments with the concentration 108 cells/ml. The surfaces were rinsed by immersion in the buffer and rotation in the shaker. The images sizes are (a, c, e) 50 µm and (b, d, f) 10 µm. The height difference is 400 nm in all images.
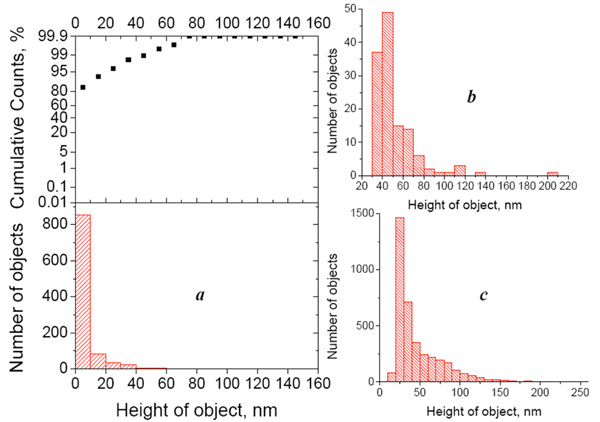
(a) Height distribution histogram (bottom) of objects detected in the control AFM image (Escherichia coli- affinitive surface exposed to the buffer) and integral probability function (top). (b) height distribution histogram of objects detected in the AFM image of the cross-control sample (Escherichia coli- affinitive surface exposed to the suspension of Bacillus subtilis fragments with the concentration 108 cells/ml). (c) Height distribution histogram of objects detected in the AFM image of the Escherichia coli- affinitive surface exposed to the suspension of Escherichia coli fragments with the concentration 108 cells/ml. All histograms were built for the particles detected from 50x50 µm2 AFM images.
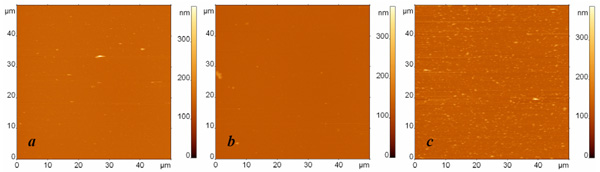
AFM images of surfaces modified with protein G and anti-Escherichia coli antibodies and exposed to (a) the buffer, (b) the suspension containing Bacillus subtilis cell fragments with the concentration 107 cells/ml, and (c) the suspension containing Escherichia coli cell fragments with the concentration 107 cells/ml. The surfaces were rinsed by immersion in the buffer and rotation in the shaker. The images sizes are 50 µm. The height difference is 400 nm in all images.
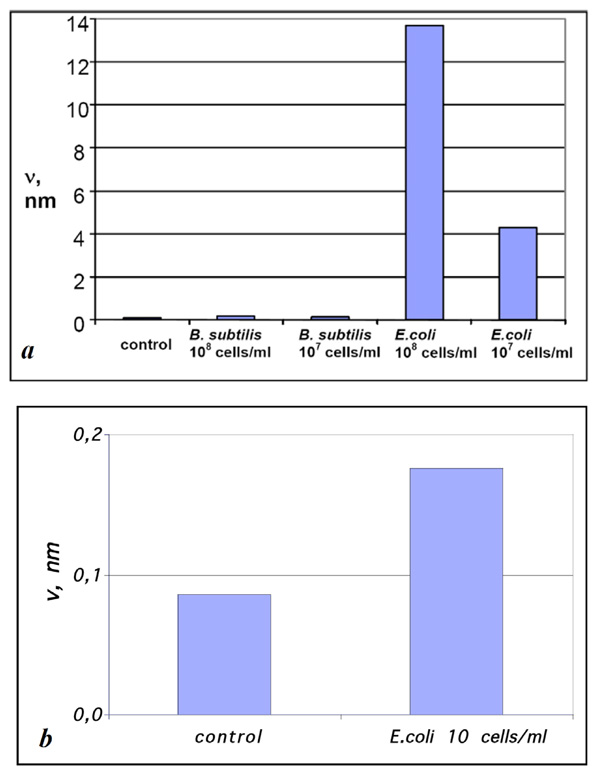
Diagrams of ν values for the samples prepared by exposing Escherichia coli – affinitive surfaces to suspensions of Bacillus subtilis and Escherichia coli fragments with concentrations (a) 108 and 107 and (b) 10 cells/ml compared with the control sample. The estimation error for ν values was below 1%.
Because our goal was to use bacterial fragments as antigens, we modified the cell destruction procedures for our purposes (see Materials and Methods). Light microscopy experiments showed that the freeze-thaw method does not disrupt microbial cells effectively, while the ultrasound treatment allows obtaining nearly complete bacterial cell destruction. We therefore used ultrasound for cell disruption in the further experiments.
We exposed the droplet of antigen suspension (analyte) in buffer to the biofunctional surface for ten minutes and then rinsed the surface by immersing the mica bed in the same buffer with subsequent shaking. The rinsing step helps to minimize the number of nonspecifically bound particles because their adhesion is weaker than the affinitive binding. Because of additional hydrodynamic flows in the surface proximity, such a rinsing method increases the efficiency of purifying the biofunctional surface from physisorbed bacterial fragments and impurities.
The specific adsorption of Escherichia coli fragments on the affinity surface is illustrated in Fig. (2), where typical AFM images of three different specimens are depicted: a droplet of buffer in the first sample (Fig. 2a, b) and droplets with bacterial fragments of Bacillus subtilis cells (Fig. 2c, d) and Escherichia coli cells (Fig. 2e, f) in the respective second and third samples were exposed to an anti-Escherichia coli antibody modified surface. In these experiments, the concentration of bacterial fragments was equivalent to 108 cells/ml. The AFM images in the left and right columns were obtained independently with the respective scan sizes of 50 µm and 10 µm. All the presented AFM images have the same scale of height contrast.
The contrast between the AFM images of the third sample (Fig. 2e, f) illustrating specific binding and the first two samples (Fig. 2a-d) illustrating the control experiment and nonspecific adsorption to the functionalized surface can be clearly seen. To characterize and compare the adsorption ability of the studied surfaces quantitatively, we developed a procedure for analyzing the quantity of bound material observed in the AFM images. Because we study the adsorption of bacterial fragments of different sizes and shapes in our experiments, the number of fragments and their height cannot be used to characterize the quantity of bound material correctly and to compare the surface binding ability adequately in different experiments. For this we can use the total volume per area v=V/S as a parameter, where V is calculated as is the volume of the ith particle in the AFM image, and S is the area of the AFM image. The value v, which has the meaning of the quantity of adsorbed material on the unit surface, serves as an indicator for estimating the presence or absence of specific binding.
In practice, to calculate the value v, we must first detect all objects in the image and calculate their volume. Both procedures (detecting the objects and calculating their volume) are integrated in the software we used. The lower height threshold of detected bacterial fragments depends on the background noise or, more precisely, on the mean purity (roughness) of the control sample (which contains buffer instead of the antigen suspension). In other words, we ignore the lower height values that are typical for the control image. Of course, the control image will never be perfectly flat, because of possible impurities in the buffer solution and imperfections of the obtained protein surfaces. In our experiments, the height distribution histogram and integral probability function showed that 95% of the observed objects are below 20 nm (Fig. 3a), and we considered only particles higher than 20 nm in the further analysis. The height distribution histograms of objects on the AFM images referring to “specific antigen” and “nonspecific antigen” samples show a significant difference (~30-fold) in the number of bound objects (Fig. 3b, c).
In the next series of experiments, we decreased the initial concentration of the analyte ten times. The resulting AFM images of specific and nonspecific adsorption together with the blank experiment are presented in Fig. (4). We again observe a significant number of specific binding events (Fig. 4c), while the number of objects adsorbed from the buffer (Fig. 4a) or from the suspension of nonspecific bacterial fragments (Fig. 4b) remains extremely low.
The values of v for the samples obtained by exposing Escherichia coli- affinitive surfaces to suspensions containing Bacillus subtilis and Escherichia coli cell fragments with the concentrations 108 and 107 cells/ml (see Figs. 2, 4) compared with control experiments are presented in Fig. (5a). The presented diagram shows a more than 10-fold increase of material bound to specific surfaces compared with the unspecific surface and “background noise.”
The results presented in Figs. (2-5) not only demonstrate the possibility of AFM visualization of single binding events between biofunctional surfaces and bacterial fragments but also indicate that the developed procedure for preparing samples allows significantly minimizing the number of physisorbed particles. Therefore, we can confirm that the developed affinity surfaces and sample preparation procedure allow obtaining a selective sensitivity of biofunctional surface and determining binding specificity by analyzing AFM images. This is very important for developing biosensor applications based on biofunctional surfaces of this type.
Unlike other surface techniques (QCM, SPR, ellipsometry, etc.) that involve analyzing the integral signal from the whole surface for detection, the sensivity of AFM as a sensor of bacterial cells is not practically limited by the sensivity of AFM itself, because antigen sizes usually considerably exceed the typical size of objects reliably detected by AFM. In other words, even a single cell adsorbed to an AFM scanning surface area will always be reliably detected. Hence, the only remaining limitation is connected with nonspecifically bound material (so-called noise), and the amount of “noise” can be minimized by appropriate protocols of biofunctional surface preparation and deposition of the analyte solution as well as by the purity of the initial reagents. In order to demonstrate the behavior of our system at extremely low concentrations of an antigen we have made the adsorption experiments of E. coli fragments from the suspension with concentration 10 cells/ml (Fig. 5b). Though the difference of v values was not so significant in this case and we cannot speak about the reliable detection, the estimation of v value was 2 times higher for specific adsorption rather than for unspecific one.
In the case of a known topography of an antigen (e.g., a particular bacterial strain), its AFM image analysis allows distinguishing an antigen from other adsorbed material and thus allows minimizing the corresponding sensivity limitation. Because our aim here was to demonstrate the system for AFM detection of bacterial fragments on biofunctional surface, we did not analyze minimization of the detection threshold of the analyte. However, our results show that there is large room for decreasing the detection threshold.
CONCLUSIONS
Using atomic force microscopy, we have demonstrated the possibility of detecting bacterial fragments in a solution for the first time. For this we developed a biofunctional surface with a selective sensitivity to a particular pathogen and elaborated a procedure for preparing specimens for AFM. We started from the conventional approach of protein G linked antibodies as a basis for the affinity surface and introduced some additional stages of treating it to meet the specific needs of preparing samples for biological AFM. We gave much attention to the premodification of the substrate in a glow discharge, the conditions of protein immobilization, and the procedures for rinsing.
To minimize unspecific binding to the biofunctional surfaces, we developed a special two-stage rinsing procedure that includes immersing the specimen in the buffer, rotating it on the shaker, and then rinsing with a water droplet.
The volume of material adsorbed onto the surface per unit area (v) can serve as a quantitative measure of specific binding. This value showed a 30-fold difference between specific and nonspecific suspensions at a concentration of 107 cells/ml and an 80-fold difference at a concentration of 108 cells/ml.
Our results demonstrate great opportunities for developing reliable AFM-based sensing procedures for detecting pathogenic bacteria, and cell detection remains possible after its complete destruction. Moreover, the possibility of visualizing single binding events offers the challenge to detect ultra-low antigen concentrations, although additional efforts should be made in this case to further minimize nonspecific binding.
ACKNOWLEDGEMENTS
Declared none.
CONFLICT OF INTEREST
Declared none.

