All published articles of this journal are available on ScienceDirect.

Regulation of clpQ+Y+ (hslV+U+) Gene Expression in Escherichia coli
Abstract
The Escherichia coli ClpYQ (HslUV) complex is an ATP-dependent protease, and the clpQ+Y+ (hslV+U+) operon encodes two heat shock proteins, ClpQ and ClpY, respectively. The transcriptional (op) or translational (pr) clpQ+::lacZ fusion gene was constructed, with the clpQ+Y+ promoter fused to a lacZ reporter gene. The clpQ+::lacZ (op or pr) fusion gene was each crossed into lambda phage. The λclpQ+::lacZ+ (op), a transcriptional fusion gene, was used to form lysogens in the wild-type, rpoH or/and rpoS mutants. Upon shifting the temperature up from 30 °C to 42 °C, the wild-type λclpQ+::lacZ+ (op) demonstrates an increased β-galactosidase (βGal) activity. However, the βGal activity of clpQ+::lacZ+ (op) was decreased in the rpoH and rpoH rpoS mutants but not in the rpoS mutant. The levels of clpQ+::lacZ+ mRNA transcripts correlated well to their βGal activity. Similarly, the expression of the clpQ+::lacZ+ gene fusion was nearly identical to the clpQ+Y+ transcript under the in vivo condition. The clpQm1::lacZ+, containing a point mutation in the -10 promoter region for RpoH binding, showed decreased βGal activity, independent of activation by RpoH. We conclude that RpoH itself regulates clpQ+Y+ gene expression. In addition, the clpQ+Y+ message carries a conserved 71 bp at the 5’ untranslated region (5’UTR) that is predicted to form the stem-loop structure by analysis of its RNA secondary structure. The clpQm2Δ40::lacZ+, with a 40 bp deletion in the 5’UTR, showed a decreased βGal activity. In addition, from our results, it is suggested that this stem-loop structure is necessary for the stability of the clpQ+Y+ message.
INTRODUCTION
The E. coli clpQ+Y+ operon, encoding an ATP-dependent protease, was originally discovered as part of the heat shock apparatus [1]. In the clpQ+Y+ operon, the first gene, clpQ+, encodes a small peptidase of 19 KDa, and the second gene, clpY+, encodes an ATPase large subunit of 50 KDa [1-3]. In the presence of ATP, the ClpYQ complex forms an active protease with an Y6Q6Q6Y6 configuration [2, 3]. Biochemical and genetic analyses have shown that the cellular proteins, SulA, RcsA, and RpoH are degraded by the ClpYQ protease [4-11], and that ClpY interacts with SulA in the yeast two-hybrid assays [12]. It was recently reported that Cpx-mediated TraJ degradation by ClpYQ is a stress-mediated response [13]. The most recent aspects for the study of the biological activity of the ClpYQ ATP-dependent protease is reviewed in Wu et al., 2008 [14].
Heat shock responses are typically observed in E. coli (reviewed in [15-17]). Upon heat shock, protein misfolding leads to a cascade of intracellular protein synthesis, usually dependent on a sigma factor, i.e., σ32, for their gene expression. The σ32 consensus binding sequences are found in the upstream region of the clpQ+Y+operon and its mRNA production is increased upon heat shock induction (Fig. 1A) [1]. However, no empirical experiment was performed to directly identify that the RpoH (σ32) is responsible for the heat shock induction of clpQ+Y+. In addition, the transcriptional start point of the clpQ+Y+ gene lies 71 bases upstream from the clpQ+ start codon (Fig. 1A). An untranslated region (UTR) upstream of this mRNA contains a 20 bp inverted repeat (IR) sequence 5’CCCCGTACTTTTGTACGGGG3’, which is unique for the clpQ+Y+ operon (Fig. 1A). In addition, from the wild bacterial genome, the 5’UTR of clpQ+Y+ also exists in other bacterial species (Fig. 1B).

(A) Nucleotide sequences of the promoter region of the clpQ+Y+ operon [1]. The transcriptional start site is shown by arrows. The consensus -10 and -35 sequences recognized by σ32 are underlined. The SD region and the start codon GTG of clpQ+Y+ are as specified. The clpQm1 carries a C to T nucleotide substitution in the region of the RpoH -10 promoter element. The dotted line indicates the deletion of 40 bps in the leader sequence in front of the translational start codon in the clpQm2Δ40::lacZ+ fusion gene. (B) The conserved regions of sequences from the 5’UTR of the clpQ+Y+ genes were aligned among the different bacterial species. The bacteria are present mainly in the Enterobacteriaceae family, i.e. Escherichia coli, Shigella flexneri, Salmonella enterica subsp. Enteritidis, Klebsiella pneumonia, Enterobacter sp., Serratia proteamaculans, Yersinia pestis, Erwinia tasmaniensis and Citrobacter koseri. The sequence that forms a hairpin structure is underlined and the IR sequence is highlighted. The Shine-Darlgarno sequence is boxed and the translational start site is in bold.
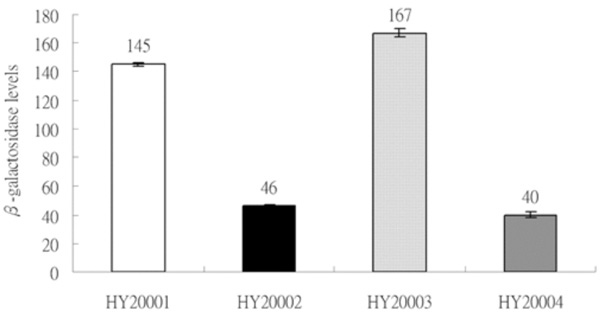
Expression of two sets of λclpQ+::lacZ gene fusion, in a transcriptional or a translational fusion type, with two different lengths of the clpQ+ promoter. The βGal activities are represented as bars, from left to right, HY20001 (op-141 bp), HY20002 (pr-141 bp), HY20003 (op-478 bp) and HY20004 (pr-478 bp). The lysogens were cultivated on LB media to log phase at 30 °C, and the enzyme assays were performed in triplicate at least three times.
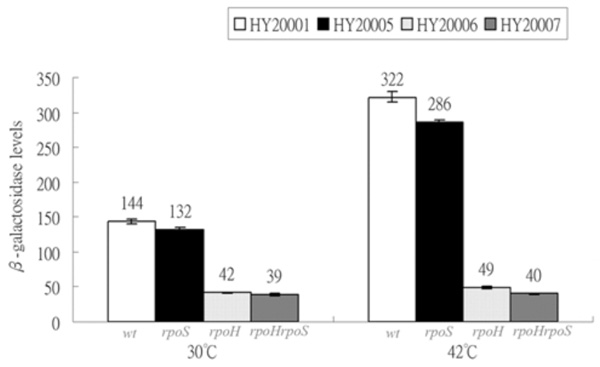
Expressions of λclpQ+::lacZ+ in the wild-type and the various sigma- factor deficient strains. HY20001 (the wild-type), HY20005 (rpoS), HY20006 (rpoH) and HY20007 (rpoH rpoS) are as indicated at 30 °C or 42 °C. The lysogens were cultivated at 30 °C first for log growth and then shifted to 42 °C for 20 min.
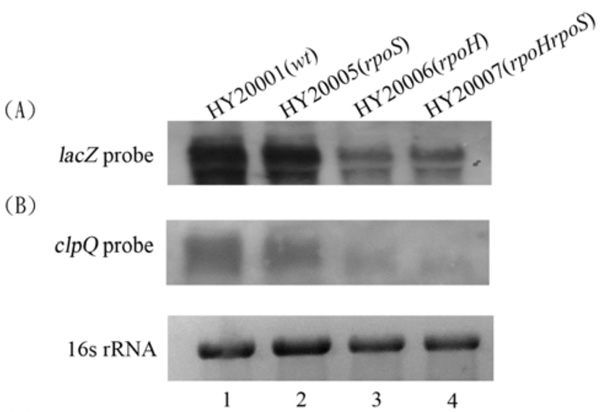
Northern blots; the total RNA were probed with a DNA fragment of lacZ + (A), or clpQ+ (B). The HY20001 (the wild-type), HY20005 (rpoH), HY20006 (ropS), and HY20007 (rpoH rpoS) mutants, each carrying λclpQ+::lacZ+, were hosts for the RNA isolation. Each lane contains 3 µg RNA for the analysis. The ethidium-bromide stained 16S rRNA was used for normalization of the gel loading.
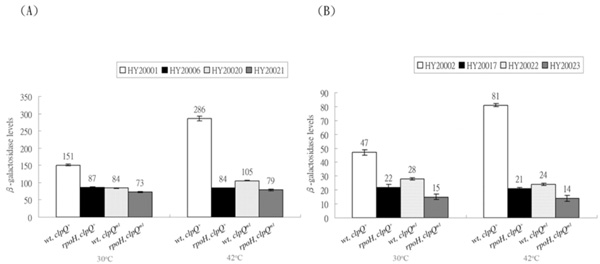
Expression of the λclpQ+::lacZ (op or pr fusion) and the λclpQm1::lacZ (op or pr fusion) in the wild-type or the rpoH mutant at 30 ° C or 42 ° C. (A) The βGal activities of HY20001 (clpQ+::lacZ+, op), HY20006 (rpoH, clpQ+::lacZ+, op), HY20020 (clpQm1::lacZ+, op), and HY20021 (rpoH, clpQm1::lacZ+, op) are as indicated. (B) In contrast, the βGal activities of HY20002 (clpQ+::’lacZ, pr), HY20017 (rpoH, clpQ+::’lacZ, pr), HY20022 (clpQm1::’lacZ, pr) and HY20023 (rpoH, clpQm1::’lacZ, pr) are also listed.
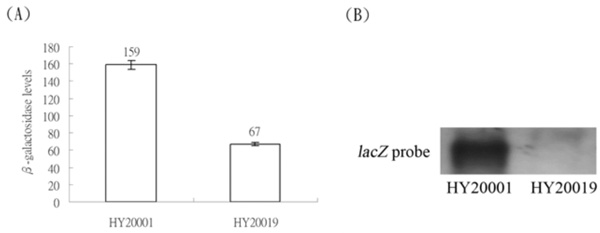
(A) Expression of the λclpQ+::lacZ+ in HY20001 and expression of λclpQm2Δ40::lacZ+, with a 40 bp deletion in the internal leader sequences, in HY20019. (B) The northern blot of the mRNAs of these two fusion genes, using the 723 bp of lacZ+ as a probe.
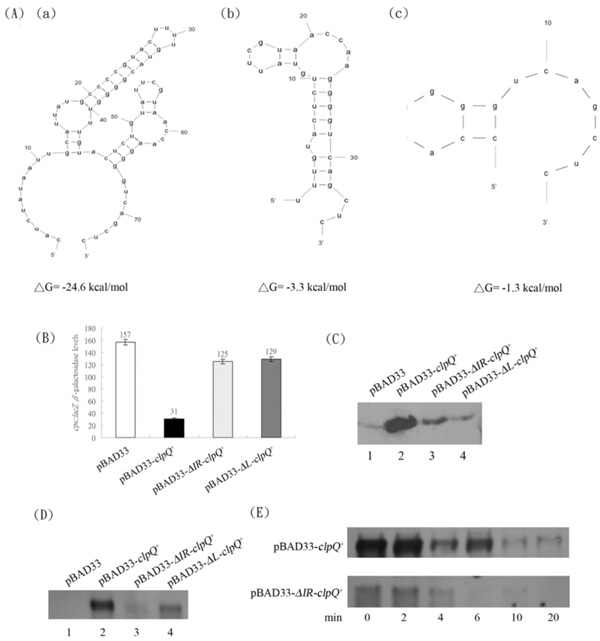
(A) The secondary structures of the 5’ UTR region of the mRNA from (a) clpQ+ with ΔG = -24.6 Kcal/mole (b) ΔIR-clpQ+ with ΔG= -3.3 Kcal/mole and (c) ΔL-clpQ+ with ΔG = -1.3 Kcal/mole. (B) The βGal of cpsB::lacZ in the presence of pBAD33, pBAD33-clpQ+, pBAD33-ΔIR-clpQ+, or pBAD33-ΔL-clpQ+, while ClpY was induced. (C) The western blot of ClpQ, expressed, in series, from the above plasmids. The loading was normalized by the readings of OD600. (D) The mRNA of clpQ+, which individually expressed from the above plasmids. The growth condition was as indicated earlier for the addition of antibiotics and 0.5 % arabinose, and the loadings were normalized by 16S rRNA (data not shown). (E) The decay of mRNAs of clpQ+ and ΔIR-clpQ+; the remaining amount of mRNA at different time points, after an addition of rifampin (at a final concentration, 200 µg/ml).

(A) The secondary structures of 5’-UTR in (a) (S-IR)-clpQ+ with ΔG = -10.1 kcal/mol and (b) (R-IR)-clpQ+ with ΔG = -24.6 kcal/mol. (B) The βGal activities of cpsB::lacZ, in the presence of ClpY, the ClpQ was induced from pBAD33-(S-IR)-clpQ+ or pBAD33-(R-IR)-clpQ+, as compared to the control pBAD33-clpQ+and pBAD33-ΔIR-clpQ+. (C) The mRNAs of clpQ+ from the pBAD33-(S-IR)-clpQ+ and pBAD33-(R-IR)-clpQ+ as compared to those from the pBAD33-clpQ+ and pBAD33-ΔIR-clpQ+.
In this study, we analyzed clpQ+Y+ mRNA production in wild-type (wt) cells versus sigma-factor deficient mutants using a λclpQ+::lacZ+ fusion gene, in which a chromosomal clpQ+Y+promoter is fused with a lacZ+ reporter. We demonstrated that the expression of the lacZ reporter gene is induced in the wild-type cells but not in rpoH-deficient mutants under the heat shock induction. The clpQm1::lacZ+ fusion gene, carrying a C to T mutation in the -10 promoter site for RpoH-binding, showed a decreased gene expression. No additional sigma factor was identified for an activation of its gene expression. Thus, through a genetic analysis, the clpQ+Y+ promoter is in vivo recognized by σ32. In addition, a stem-loop structure (IR) at the 5’ end of the UTR of clpQ+Y+ mRNA is shown to be necessary for clpQ+Y+ gene expression.
MATERIALS AND METHODOLOGY
Materials and Media
Bacterial strains and phages used in this study are listed in Table 1. LB complex media was purchased from Difco and the bacteria were grown in LB broth, with an addition of the appropriate antibiotics as required. Supplements were added as the following final concentrations when needed: 100 μg ampicillin (Ap) ml-1; 40 mg X-gal ml-1. The Taq DNA polymerase, restriction endonucleases, and other enzymes were obtained from Takara, New England BioLabs and Biolab (Taipei). Chemicals were obtained from Sigma or Wako.
The E. coli Strains and Phages Used in this Study
| Strains or Phages | Description | References or Derivation |
|---|---|---|
| Strains | ||
| MC4100 | F-araD139Δ(argF-lac)U169 rpsL150 relA1 fibB5301 deoC1 ptsF25 rbsR | [18] |
| KY1429 | MC4100 rpoH6(Am) zhf-50::Tn10 | [19] |
| RH90 | MC4100 rpoS-359::Tn10 | [20] |
| SM25 | MC4100 rpoH rpoS | [21] |
| HY20001 | MC4100 λclpQ+::lacZ+ (op-141 bps) | This work |
| HY20002 | MC4100 λclpQ+::’lacZ (pr-141 bps) | This work |
| HY20003 | MC4100 λclpQ+::lacZ+ (op-478 bps) | This work |
| HY20004 | MC4100 λclpQ+::’lacZ (pr-478 bps) | This work |
| HY20005 | rpoS λclpQ+::lacZ+ (op-141 bps) | This work |
| HY20006 | rpoH λclpQ+::lacZ+ (op-141 bps) | This work |
| HY20007 | rpoH rpoS λclpQ+::lacZ+ (op-141 bps) | This work |
| HY20017 | rpoH λclpQ::’lacZ (pr-141 bps) | This work |
| HY20019 | MC4100 λclpQm2Δ40::lacZ+ (op) | This work |
| HY20020 | MC4100 λclpQm1::lacZ+ (op-141 bps) | This work |
| HY20021 | rpoH λclpQm1::lacZ+ (op-141 bps) | This work |
| HY20022 | MC4100 λclpQm1::’lacZ (pr-141 bps) | This work |
| HY20023 | rpoH λclpQm1::’lacZ (pr-141 bps) | This work |
| AC3112 | lon clpQY cpsB::lacZ | [10] |
| Phages | ||
| λRS45 phage (Φ) | imm21 ind+ bla’-lacZsc | [22] |
| λclpQ+::lacZ+ (op-141 bps) | carrying clpQ+Y+ promoter (141 bps) with lacZ+ | This work |
| λclpQ+::’lacZ (pr-141bps) | carrying clpQ+Y+ promoter (141 bps) with’lacZ | This work |
| λclpQ+::lacZ+ (op-478 bp) | carrying clpQ+Y+ promoter (478 bps) with lacZ+ | This work |
| λclpQ+::’lacZ (pr-478) | carrying clpQ+Y+ promoter (478 bps) with’lacZ | This work |
| λclpQm1::lacZ+ (op-141) | carrying a C to T mutation in -10 promoter (op) | This work |
| λclpQm1::’lacZ (pr-141) | carrying a C to T mutation in -10 promoter (pr) | This work |
| λclpQm1Δ40:: lacZ+ (op) | carrying a 40 bp internal deletion in the 5’UTR (op) | This work |
The Plasmids Used in this Study
| Plasmids | Description | References |
|---|---|---|
| pRS415 | bal-Tl4-EcoRI-SmaI-BamHI-lacZ+ | [22] |
| pRS414 | bal-Tl4-EcoRI-SmaI-BamHI lacZ’ | [22] |
| pWPC80 | pACYC184 with clpQ+Y+ and its surrouding sequences | [9] |
| pWF1 | pACYC184(Tetr)-clpQ+clpY+ | [9] |
| pSY1 | clpQ+Y+ promoter (141 bps)-lacZ+ in pRS415 | This work |
| pSY2 | clpQ+Y+ promoter (141 bps)-lacZ’ in pRS414 | This work |
| pSY3 | clpQ+Y+ promoter (478 bps)-lacZ+ in pRS415 | This work |
| pSY4 | clpQ+Y+ promoter (478 bps)-lacZ’ in pRS414 | This work |
| pSY5 | clpQm1::lacZ+ in pRS415 | This work |
| pSY6 | clpQm1::lacZ’ in pRS414 | This work |
| pSY7 | pRS415-clpQm2Δ40::lacZ+ | This work |
| pSY8 | pRS414-clpQm2Δ40::lacZ’ | This work |
| pBAD33 | ori (pACYC184, Cmr) araC PBAD | [26] |
| pSY9 | pBAD33-clpQ+ | This work |
| pSY10 | pBAD33-ΔIR-clpQ+ | This work |
| pSY11 | pBAD33-ΔL-clpQ+ | This work |
| pSY12 | pBAD33-(S-IR)-clpQ+ | This work |
| pSY13 | pBAD33-(R-IR)-clpQ+ | This work |
| pBAD24 | ori (pBR322, Ampr) araC PBAD | [26] |
| pSY14 | pBAD24-clpY+ | This work |
P1 Transduction and Other General Methods
P1vir, λ derivative phages, and transduction were prepared as described by Silhavy et al. [23]. Mutant strains were constructed by P1 transduction by procedures as described by Miller [24]. For plasmid constructions, E. coli XL-1 blue competent cells were used. Plasmid transformations were performed as described by Chung et al. [25]. Plasmid purification was performed according to the instructions of manufacturers (Viogene).
Primers, Construction of Plasmids and DNA Sequencing
The forward (F) and reverse (R) primers used in this study were listed below with the restriction enzyme site underlined and the substituted nucleotides bold. F1: 5'-CGGAATTCCCGGGGGTTGAAA-3', F2: 5'-CCGGAATTCAGCCGTACCAGGATCTGCTGCAAACTCCTG-3', F3: 5'-CCGAATTCCCGGGGGTTGAAACCCTCAAAATCCCCCTCATCTATAATTGCATTATGCCCCGTA-3', F4: 5'-CCATCTATAATTGCATTATGCCAAGGGGTCAGCT-3', F5: 5'-GGGGTACCCATCTATAATTGCATTATGCCC-3', F6: 5'-GGGGTACCTTTGTACTCTGTATTCGTAAC-3', the F7: 5'-GGGGTACCCCAAGGGGTCAGCTCGTGACAA-3', F8: 5'-GGGGTACCCATCTATAATTGCATTATGCCCCGTACTTTTTTTTTTTTTTTGTACTCTGTATTCGTAACCAAGGGGTCAG-3', F9: 5’-GGGGTACCCATCTATAATTGCATTATGGGGGCATGTTTTCATGCCCCTTTGTACTCTG TATTCGTAACCAAGGGGTCAG-3’, R1: 5'-GCGGGATCCACATGGCCGTTACGGC-3', R2:5'-CATAATGCAATTATAGATGG-3', R3: 5'-CGCGGATCCGCATCCGCAGTACCGCCCGCAAAG-3'. R4: 5'-CCCAAGCTTTTACGCTTTGTAGCTTAATT-3'. All the plasmids, used in this study, were listed in Table 2. Plasmids pSY1 [clpQ+Y+ promoter (141 bps)-lacZ+, in pRS415], pSY2 [clpQ+Y+ promoter (141 bps)-lacZ’ in pRS414], pSY3 [clpQ+Y+ promoter (478 bps)-lacZ +, in pRS415] and pSY4 [clpQ+Y+ promoter (478 bps)-lacZ’, in pRS414] were constructed as follows. The plasmid, pWPC80 [9], was used as a template for an amplification of a promoter region of clpQ+Y+. The PCR was used to amplify the genes surrounding the upstream region, including the clpQ+Y+ promoter, the Shine-Dalgarno (SD) site, and the first thirteen amino acids of clpQ+, and the resulting DNA fragment was fused in-frame to lacZ’ in pRS414 [22], a translational lacZ’ fusion (pr) plasmid [22]; this upstream region was also cloned separately in front of lacZ+ in pRS415 plasmid with a transcriptional lacZ+ fusion (op). Two sets of primers were used. Set 1: F1 and R1. Set 2: F2, R1. The PCR conditions were performed according to the manufacturer’s instructions, and then the PCR products were purified with Viogene Gel-M kits. Both pRS414 and pRS415 plasmids were cut with EcoRI and BamHI and ligated with the DNA fragments treated with the same restriction enzymes. The resulting ligated mixtures were then transformed into the E. coliXL-1 blue, and the transformants were selected on ampicillin agar plates. Plasmids isolated from colonies grown on the selective media were subjected to restriction enzyme analysis, followed by DNA sequencing. To construct clpQm1::lacZ+, with a C to T mutation, a set of primers was used as follows: F3 and R1. Similar procedures as described above were used to clone the PCR fragment carrying clpQm1 upstream of lacZ. Two resulting plasmids, clpQm1::lacZ+ in pRS415 and clpQm1:: lacZ’ in pRS414, were designated pSY5 and pSY6, respectively. Several PCR steps were then used to construct a clpQm2Δ40:: lacZ+, with a 40 bp deletion in the internal region of UTR upstream of the fusion gene. To construct a clpQm2Δ40 mutation, a two-step PCR was used to amplify the clpQ+ promoter and its adjacent region. The upstream region primers were F2 and R2. The downstream primers were F4 and R3. Two overlapping PCR fragments were then used as the template and were amplified by the primers of F2 and R3. The resulting DNA fragment with a 40 bp deletion within the leader sequences (as shown in Fig. 1A) was cloned into pRS415 and pRS414 at EcoRI-BamHI sites, and the resulting plasmids were designated pRS415-clpQm2Δ40::lacZ+ (pSY7) and pRS414-clpQm2Δ40:: lacZ’ (pSY8), respectively.
Plasmids pBAD33-clpQ+ (pSY9), pBAD33-ΔIR-clpQ+ (pSY10) and pBAD33-ΔL-clpQ+ (pSY11) were constructed as described below. Using the pWF1 [9] as the template, the various DNA fragments surrounding the upstream region of clpQ+ gene and the upstream region alone were separately PCR-amplified. The resulting DNA fragments were cloned in series into the pBAD33 at the KpnI-HindIII sites, and the cloned genes were all under the pBAD promoter control. Consequently, the plasmid pBAD33-clpQ+carries the upstream 71 nucleotides from the translational start site of clpQ+ and clpQ+ itself. The F5 and R4 primers were used. The plasmid pBAD33-ΔIR-clpQ+, which contains 35 nucleotides upstream of the GTG initiative codon of clpQ+ and clpQ+ itself under the pBAD promoter, was constructed, using F6 and R4 primers to amplify a DNA fragment of ΔIR-clpQ+. The plasmid pBAD33-ΔL-clpQ+ was constructed, in which the 15 nucleotides upstream of the first GTG codon and clpQ+ itself were under the pBAD promoter control. To generate ΔL-clpQ+, F7 and R4 primers were used. In addition, the plasmid pBAD33-(S-IR)-clpQ+ (pSY12) was constructed, which carries the substituted T sequences at the 5’ end of the UTR of clpQ+ (indicated with bold in the primer F8), using the F8 and R4 primers. Plasmid pBAD33-(R-IR)-clpQ+ (pSY13) carries the substituted nucleotides at the 5’ end of UTR of clpQ+, resulting in a newly introduced secondary structure. The F9 and the R4 primers were used for generating the DNA fragment of (R-IR)-clpQ+. The plasmid pBAD24-clpY+ (pSY14) was constructed as follows. Using the pWF1 as the template, the clpY+ was amplified, and the resulting DNA fragment was cloned into the pBAD24 at the EcoRI-HindIII cutting sites [10, 26]. All the constructed plasmids with the correct DNA insert were identified by restriction enzyme mapping as well by DNA sequencing.
Construction of λclpQ::lacZ Fusion Gene
Plasmids pSY1 (clpQ+:: lacZ+, op-141), pSY2 (clpQ+::lacZ’, pr-141), pSY3 (clpQ+:: lacZ+, op-478) and pSY4 (clpQ+::lacZ’, pr-478) were made. These four plasmids were transformed into the E. coli XL-1 blue, and the resultant transformants (Apr) were selected on ampicillin plates. The transformant cells were then infected with the lambda derivative λRS45 and selected for blue plaques on X-gal plates leading to clpQ+::lacZ+ transcriptional or clpQ+::’lacZ translational fusion genes transferred into λRS45Ф [22]. The derivative λRS45ФclpQ::lacZ+ (or λclpQ+ ::’lacZ) fusion gene was made with different upstream length of the clpQ+Y+ promoter. Each phage lysate was used to infect E. coli and the resultant single phage lysogen was selected. A single phage lysogen was identified by measurement of βGal levels of five lysogens, and those with the lowest expression value were selected as the single lysogens.
The βGal Assays
The βGal activities were assayed as described by Miller [24]. The cells were grown to log phase, and activity was measured by the βGal assay. All of the βGal activity was determined in triplicate for each assay. Each analysis was repeated at least three times.
Northern Hybridization Assays
The cells were grown in 5 ml of broth to exponential phase at 30 oC (OD600 about 0.5). At each time point, total RNA was isolated by Qiagene RNeasy Mini kits. Equal amounts of total RNA (3 μg each) were separated under denaturing conditions in a 1% agarose-2.1 M formaldehyde-MOPS [morpholinopropane-sulfonic acid] gel, stained with ethidium bromide, and blotted onto nitrocellulose (Schleicher & Schuell) with 20X SSC. The DNA probes were labeled with DIG-dUTP by random priming (Roche). The DNA fragments used for probes were obtained by PCR amplification. The 401 bp DNA fragment corresponding the clpQ+ structural gene was used as a probe for clpQ+ mRNA, and a 723 bp DNA fragment containing a lacZ+ gene was used as a probe for the clpQ+::lacZ+ mRNA. The membrane was hybridized with the appropriate probe and washed. The signals were produced by an addition of the anti-DIG antibody with fluorescent alkaline phosphatase conjugate and CDP-star (NEB BioLabs) to the membrane and visualized by an exposure to an X-ray film.
The cpsB::lacZ Assays and the Western-blot Analysis for ClpQ
Cells, AC3112 (lon, clpQY, cpsB::lacZ) [10] carrying plasmids pBAD33-clpQ+, pBAD33-ΔIR-clpQ+, pBAD33-ΔL-clpQ+, pBAD33-(S-IR)-clpQ+, or pBAD33-(R-IR)-clpQ+ in combination with pBAD24-clpY+ plasmids were grown overnight on LB with ampicillin (50 μg/ml) and chloramphenicol (12.5 μg/ml) with an addition of arabinose (0.5 %) to induce the PBAD promoter. The overnight cultures were inoculated 1:100 into fresh media with the above indicated ingredients and were grown into log phase, and then half of the cells were immediately subjected to the βGal assays for the cpsB::lacZ expression. The other half of the log cells were harvested and subjected to the western blot analysis. The polyclonal ClpQ antibody was used to detect the ClpQ protein, which was separately encoded by the above three plasmids pBAD33-clpQ+, pBAD33-ΔIR-clpQ+ and pBAD33- ΔL-clpQ+. The procedure was adopted as described previously [12]. The western blots were developed by the enhanced chemiluminescence (Pierce).
RESULTS
The Expression of clpQ+::lacZ Fusion Gene in λRS45
To monitor the expression of the clpQ+Y+ genes in E. coli, two sets of clpQ+::lacZ fusion genes were made; in each one, the clpQ+Y+ promoter is in front of lacZ. Both fused genes have the lacZ gene under the clpQ+Y+ promoter control, but they carry the different length of the upstream sequence in either a transcriptional or a translational fusion type. After transferring these four clpQ+::lacZ fusion gene into the λRS45, the resulting phages, λclpQ+::lacZ+ (op-141 bp), λclpQ+::’lacZ (pr-141 bp), λclpQ+::lacZ+ (op-478 bp), and λclpQ+::’lacZ (pr-478 bp), were separately lysogenized into a lacZ-deficient strain (MC4100). These four newly lysogens were isolated, namely, HY20001, HY20002, HY20003 and HY20004, correspondingly. The βGal activity was then measured for the each single phage lysogen. Despite the two different sizes of the clpQ+Y+ promoter fragments (141 bps or 478 bps), the two lysogens in a transcriptional type (HY20001 and HY20003) had an almost equal βGal activity of lacZ gene fusion (Fig. 2); similarly this was observed in a translational type (HY20002 and HY20004) (Fig. 2). Thus, only one set of lysogens (the op and pr lacZ fusion gene with an identical length of the upstream clpQ+Y+ promoter) would be used for the further analysis.
ClpQ+Y+ Promoter-Mediated Basal and Heat Shock-Induced Gene Expression Requires RpoH
It was shown that clpQ+Y+ promoter has the RpoH (σ32)-binding site [1]. However, a putative -10 site for an RpoS(σ38)-binding was partially overlapped with the similar RpoH recognition site (Fig. 1A). Therefore, to further identify that RpoH and/or RpoS have effects on clpQ+Y+ gene expression, the λclpQ+::lacZ+ (op-141 bp) was lysogenized into the wild-type strain MC4100 [18], and mutant strains KY1429 (rpoH) [19], RH90 (rpoS) [20], and the double mutant SM25 (rpoH rpoS) [21]. The four resulting lysogens all carried λclpQ+::lacZ+ (op-141 bp) as a prophage. The basal and heat induction of the clpQ+::lacZ+ gene expression were measured among these four lysogens: HY20001 (the wild-type), HY20005 (rpoS), HY20006 (rpoH) and HY20007 (rpoS rpoH). HY20001 and HY20005 have identical βGal activity at 30 oC (Fig. 3). In contrast, HY20006 (rpoH) and HY20007 (rpoS rpoH) show a decreased βGal activity (Fig. 3). At 42 oC, the wild-type HY20001 and the HY20005 (rpoS) mutant have increased βGal activity, whereas HY20006 (rpoH) and HY20007 (rpoS rpoH) maintain a basal expression (Fig. 3). Thus, RpoH, but not the RpoS, appears to control the clpQ+::lacZ+ gene expression, and the clpQ+::lacZ+ expression increases under the heat induction.
The mRNA Transcripts of clpQ+::lacZ+ and clpQ+Y+ in Wild-Type and RpoH/RpoS Mutants
Northern blot analyses were used to demonstrate that the clpQ+::lacZ+ mRNA expression is well correlated with its βGal levels in the above bacterial strains. Total RNA was isolated from the wild-type cells (HY20001), rpoS (HY20005), rpoH (HY20006), and rpoH rpoS (HY20007) double mutants at 30 oC. Using a lacZ+ probe, significant RNA transcripts of clpQ+::lacZ+ were observed in the wild-type and the rpoS mutant, whereas only a minor transcript was detected in the σ32-deficient mutant, HY20006, and in the double mutant rpoS rpoH, HY20007 (Fig. 4A). Similar results were observed using a clpQ+ probe to detect the clpQ+Y+ mRNA production among the wild-type and the various sigma-factor deficient strains (Fig. 4B).
Expression of clpQm1::lacZ, with a C to T Mutation in the -10 RpoH-biding Site
To reversely show that a specific mutation at the promoter region of clpQ+Y+ also has an effect on its gene expression, a C to T cis-mutation in the -10 promoter region was made in both pRS414-clpQm1::lacZ’ and pRS415-clpQm1::lacZ+ plasmids. The clpQm1 has a nucleotide substitution, 5’C~CCTCATCT3’, in the -10 promoter region distinct from the consensus sequence for the RpoH-binding, 5’C~CCCCATCT3’ (Fig. 1). The two promoter mutant plasmids carrying clpQm1::lacZ (op or pr fusion) were separately transferred into λRS45, and the resulting phages were designated λclpQm1::lacZ+ (op) and λclpQm1::’lacZ (pr), respectively. These two phages were then lysogenized into the wild-type and the rpoH mutant, and the four resultant lysogens were HY20020 (op), HY20021 (op, rpoH), HY20022 (pr), and HY20023 (pr, rpoH). As a control, a lysogen that carries the wild-type λclpQ::’lacZ (pr-141 bps) in the rpoH background was also made and named HY20017. The βGal activity was then measured for all the single phage lysogens. As shown, the expression of clpQm1::lacZ (op or pr fusion) was decreased 1.7-fold as compared to that of the wild-type λclpQ+::lacZ (Fig. 5) [compare HY 20020 to HY20001 (op) at 30 oC (Fig. 5A), and HY20022 to HY20002 (pr) at 30 oC in (Fig. 5B)]. As shown before, the wild-type clpQ+::lacZ (op or pr fusion) increased its expression at 42 oC [see HY20001 at 42 oC and at 30 oC (Fig. 5A) and HY20002 at 42 oC and at 30 oC in (Fig. 5B)]. But the gene fusion was not activated in the rpoH mutant (see HY20006 at 30 oC and at 42 oC and HY20017 at 30 oC and at 42 oC ). However, clpQm1::lacZ (op or pr fusion) was not highly induced at 42 oC [see HY20020 at 42 oC and at 30 oC (Fig. 5A) and HY20022 at 42 oC and at 30 oC (Fig. 5B)]. In addition, in the rpoH mutant, clpQ m1::lacZ (op or pr fusion) was expressed at a basal or at a slightly lower level than its expression in the wild-type cells [(Fig. 5A) and (Fig. 5B), compare HY20021 to HY20020 and compare HY20023 to HY20022, at 30 oC or 42 oC]. Thus, clpQm1, carrying a C to T mutation, has a negative effect on the clpQ+Y+ promoter activity, and it is not activated by the RpoH.
Expression of clpQm2Δ40::lacZ+, with a Deletion of 40 bps in the 5’UTR
The first transcriptional start site of the clpQ+Y+ operon was mapped 71 bp upstream from the translational start site. An UTR is present upstream from an initiation codon for the clpQ+Y+ operon. It was logically speculated that this UTR might have an effect on clpQ+::lacZ+ gene expression. To test this possibility, a clpQm2Δ40bp mutation, with a 40 bp deletion of the internal region of the UTR, was constructed in the lacZ+ gene fusion [see (Fig. 1)]. This deletion mutant, clpQm2Δ40::lacZ+, a transcriptional fusion type, was crossed into the λ phage, and the resulting phage λRS45clpQm2Δ40::lacZ+ (op) was lysogenized into the wild-type cells, MC4100. The newly formed lysogen, HY20019, displayed a significant decrease in βGal activity, consistent with its mRNA production by northern blot analysis (Fig. 6). In addition, the clpQm2Δ40bp::’lacZ (pr) mutant in a protein fusion construct (pRS414) had much lower βGal activity, and no blue plaques could be selected on the X-gal plates when crossed into the λ phage (data not shown).
Functional Activity of Inverted Repeat Sequence in the 5’ UTR of clpQ+Y+
Most single strand RNA form a secondary structure. Utilizing an in silico M-Fold program (http://frontend.bioinfo.rpi.edu/applications/mfold/) [27], a predicted stem-loop structure, including the IR sequence, with a calculated ΔG = -24.6 kcal/mol was identified in the untranslated leader region (UTR) upstream of the clpQ+Y+ structural gene (Fig. 7A-a). To demonstrate that the stem-loop structure is necessary for the expression of clpQ+Y+, DNA fragments with different lengths of the leader sequence upstream of clpQ+ were cloned into plasmid pBAD33, and all of the clpQ+ constructs, with different upstream sequences, were under the heterologous pBAD promoter control (induced by arabinose). Three different plasmids were constructed and designated pBAD33-clpQ+, pBAD33-ΔIR-clpQ+ and pBAD33-ΔL-clpQ+, and the secondary structures of the 5’ regions of their clpQ+ mRNA were predicted (Fig. 7A-a, b, c.) To evaluate the physiological activity of the above three plasmids encoded ClpQ(s), we used the AC3112 strain for an assay of a functional ClpQ. Since AC3112, a lon clpQY mutant, carries a cpsB::lacZ, it is expressed a higher βGal activity due to a stable activator, RcsA [9-11]. When the AC3112 carries both pBAD33-clpQ+ and pBAD24-clpY+ plasmids, it has a decreased βGal activity of the cpsB::lacZ, due to an instability of RcsA, targeted by the ClpYQ under the arabinose induction. AC3112 cells that carry pBAD33 and pBAD24, as a negative control, had higher βGal activities of cpsB::lacZ. However, while cells carrying plasmids pBAD33-ΔIR-clpQ+ or pBAD33-ΔL-clpQ+, in the presence of pBAD24-clpY+, each bacterial strain had much higher βGal activity from its cpsB::lacZ (Fig. 7B). Western blot analysis was then used to show that ClpQ was accumulated lower in the cells carrying plasmids pBAD33-ΔIR-clpQ+ or pBAD33-ΔL-clpQ+, as compared to the cells carrying the wild-type pBAD33-clpQ+ (Fig. 7C). To further identify that the instability of clpQ+ mRNA leads to a loss of the functional ClpQ, a northern blot analysis was used to detect the clpQ+ messages in all of the above strains. As shown, the mRNA levels of ΔIR-clpQ+ or ΔL-clpQ+ were much lower than those of clpQ+ (Fig. 7D). In addition, the stability of the wild-type clpQ+ and ΔIR-clpQ+ mRNAs was measured at different time points after the addition of rifampin to the culture media to block the re-initiation of transcription. Results show that the message of the wild-type clpQ+ is much more stable than that of the ΔIR-clpQ+ (Fig. 7E).
The Stem-loop Secondary Structure (IR) of 5’UTR of clpQ+Y+ Responsible for its mRNA Stability
To further verify that a stem-loop structure at the 5’ end of the leader sequences is responsible for the stability of clpQ+ mRNA, the substituted nucleotides were placed at the 5’UTR with or without a disruption of its stable secondary structure. As described earlier, the two new plasmids, pBAD33-(S-IR)-clpQ+ and pBAD33-(R-IR)-clpQ+, were constructed. In these constructs, the (S-IR)-clpQ+ gene has replaced the nucleotides on the 5’ end of UTR, with a ΔG= -10.1 kal/mol, that destabilized a stem-loop structure (Fig. 8A-a); in contrast, the pBAD33-(R-IR)-clpQ+ gene has replaced the nucleotides on the 5’ end of UTR, with a lower ΔG= -24.6 kal/mol, that stabilized a stem-loop structure. AC3112 cells, which carry pBAD24-clpY+ and pBAD33-(S-IR)-clpQ+, show a higher βGal activity, as compared to that of cells carrying pBAD24-clpY+and pBAD33-clpQ+ (Fig. 8B). In contrast, cells that carry pBAD24-clpY+and pBAD33-(R-IR)-clpQ+ show the decreased expression of cpsB::lacZ (Fig. 8B). AC3112, which carries pBAD33-ΔIR-clpQ+ and pBAD24-clpY+, has higher βGal activity of the cpsB::lacZ expression as that of cells with pBAD33 and pBAD24 (Fig. 8B). In the northern blot analysis, the level of (S-IR)-clpQ+ message was much lower than that of the clpQ+ and was more similar to that of the ΔIR-clpQ+; in contrast, the message of (R-IR)-clpQ+ was similar to that of the wild-type clpQ+ (Fig. 8C).
DISCUSSION
Using a clpQ+::lacZ, a gene fusion, the results presented here show that clpQ+::lacZ has similar βGal activity in constructs containing either a long or a short region of the upstream sequence of the clpQ+Y+ promoter. We have shown that the further sequence upstream of the clpQ+Y + promoter has no significant effects on its gene expression. Yet, as usual, a higher βGal level was detected with the clpQ+::lacZ+ transcriptional fusion as compared to the translational fusion, and it is accordingly due to a stronger SD region of the LacZ+ itself in the transcriptional gene fusion [22].
Coincidentally, clpQ+Y+ and clpQ+::lacZ (op or pr fusion) genes have increased expression when the temperature is up-shifted from 30 oC to 42 oC. Little of clpQ+Y+ or clpQ+::lacZ+ mRNA was observed in the rpoH mutant. In addition, a clpQm1::lacZ+, with a C to T mutation in the RpoH -10 consensus sequences, was shown to have decreased βGal activity, independent of a functional RpoH; there was also no heat shock induction in clpQm1::lacZ (op or pr fusion). Using the transcriptional start site as a primer initiating site in the RT-PCR, the end-product for the transcripts of clpQm1::lacZ+, was less accumulated as compared to those of the wild-type clpQ+::lacZ+ (data not shown). However, no signal was detected for the transcripts of both fusion genes, while using an annealing primer complementary to the nucleotides upstream of the transcriptional start site (data not shown). These results indirectly implied that both clpQ+::lacZ+ and clpQm1::lacZ+ should have the identical transcriptional start site. In addition, we also made an additional mutation in the clpQ+Y+ promoter region at the possible -10 site for the RpoS binding [see in the (Fig. 1)], and this mutation has no effects on an activity of the clpQ+Y+ promoter (data not shown). Therefore, we have experimentally shown that clpQ+Y+ gene expression is mediated by the RpoH factor.
The deletion of the IR or UTR in the ΔIR-clpQ+, ΔL-clpQ+ and the clpQm2Δ40::lacZ gene fusion leads to decreased clpQ+ mRNA. In addition, the mRNAs of ΔIR-clpQ+ mutants, without a stem-loop region at the 5’-UTR, decayed much faster. However, no significant sequences were observed in the 5’ UTR of the clpQ+Y+ messages acting as a signal for targeting by the RNase E degradosome (an AU rich or A/GAUU/AU) [28, 29] or the RNase III (A/UNAGA/UG) [30]. We also found that a functional ClpQ is not affected by a higher temperature (data not shown); thus, there is no thermo-sensitive segment in the clpQ+ message. However, while obliterated the IR in the 5’ UTR of clpQ+Y+ mRNA but retained its stem-loop structure, a stable mRNA was present. In E. coli, it has been reported that the 5’ stem-loop structure stabilizes the mRNA of papA (pyelonephritis-associated pili gene) [31] and ompA (outer membrane protein) [32, 33] under physiological conditions, and it was later found that ompA is being regulated by the small micA RNA [34]. Currently, a model is emerging of small RNA sequences that are involved in gene regulation [35]. In some cases, an Hfq (a host factor for RNA phage Qβ phage replication) [36], RNA-binding protein is also involved in it [35]. It stabilizes sRNAs against decay and also helps an association with their mRNA target [35]. However, no such effects were found for the clpQ+::lacZ+ fusion gene expression (our own data). Besides, in the wild bacterial genome, the 5’ UTR was also found in the clpQ+Y+ mRNA transcripts mainly in the Enterobacteriaceae family. These bacteria all have the conserved sequences of the 5’ UTR of clpQ+Y+ mRNA for a predicted secondary stem-loop structure. However, via the bioinformatics, the conservation of sequence of the small RNA is not sufficient for a searching of a regulation of the target gene; a more sophisticated algorithm is needed [37]. It is noteworthy that this is the first example in the ATP-dependent proteases to demonstrate that the 5’ stem-loop structure itself participates in the stability of its downstream mRNA. It is of interest to determine whether this stem-loop structure in the 5’UTR plays a role for its gene expression under certain physiological condition and also whether there is a regulation of clpQ+Y+ by the other regulatory proteins or via the small RNA.
ACKNOWLEDGEMENTS
We thank Dr. Hengge-Aronis, Dr. Ramos, Dr. Yura, and Dr. M. Kanemori, for bacterial strains. We thank Dr. R. F. Liu for technique supports. We also thank Dr. S. Gottesman for the comments. Thanks to Dr. B. Collins for a support. This work was supported by grants from the Department of Health (DOH89-TD-1068, DOH90-TD-1078, DOH-91-TD-1082) and National Science Council (NSC93-2313-B-002-077) of Taiwan, R.O.C.

