All published articles of this journal are available on ScienceDirect.

Cyclooxygenase-2 Inhibition Enhances Activation of T Helper Type 1 Responses During Salmonella Infection
Abstract
Production of IL-12 and IFN-γ secretion are important components of the protective host response against the intracellular bacterial pathogen, Salmonella typhimurium. While infection with Salmonella does elicit this T helper type 1 response, its magnitude does not appear to be sufficient to prevent infection or limit pathogenesis. Therefore we have investigated factors which might limit a T helper type 1 response following infection. Previously we found that infection of antigen presenting cells with Salmonella dramatically increases cyclooxygenase-2 (COX-2) activity, resulting in high levels of prostaglandin E2 (PGE2). Since PGE2 production can have profound effects on initiation of T helper type 1 responses, we questioned whether this mediator might limit antigen-specific T cell activation. Here we show that blockage of COX-2 activity with the selective inhibitor celecoxib leads to enhancement of the T helper type 1 components stimulated by Salmonella infection. In vitro studies demonstrate the induction of IL-12 and IFN-γ upon Salmonella exposure, which are further increased following COX-2 inhibition. Taken together these in vitro studies suggest that COX-2 activity can limit a salmonella-initiated T helper type 1 response.
INTRODUCTION
Oral inoculation of susceptible mice with Salmonella typhimurium results in a disease state similar to human typhoid fever and serves as a useful model for intracellular bacterial infection. During infection of the intestinal mucosa, Salmonella organisms invade and then survive and grow within resident macrophages and dendritic cells [1, 2]. If the inoculum is sufficient, a lethal, systemic disease ensues. Salmonella’s capacity for intracellular survival makes the initiation of an effective cell-mediated immune response crucial for survival of the host [3-6]. Fundamental to this response is the activation of macrophages and dendritic cells to produce IL-12 p70, eliciting IFN-γ production from CD4+ T cells, which further enables infected cells to kill the pathogens they harbor [7, 8]. The requirement for cellular immunity is observed in humans with genetic defects in IL-12 receptor [3, 9] or IFN-γ receptor [7] signaling, who frequently present with recurrent Salmonella infections. In the animal model, deletion or antagonism of IL-12 [6] or IFN-γ [10, 11] increases susceptibility to salmonellosis, further supporting the importance of these components in resolving infection. While these cytokines are certainly produced in response to infection [6, 12], the fact that in vivo treatment of mice with recombinant IL-12 augments protection [13] suggests that endogenous IL-12 production may be less than optimal.
Although the presence of IL-12 at the time of T cell activation facilitates the development of the T helper cell response, it has been suggested that another player, PGE2, can influence this process. Specifically, the ratio of IL-12 to PGE2 is the limiting factor in initiation of IFN-γ production and commitment to a T helper type 1 lineage [14]. PGE2 has been shown to inhibit the secretion of IL-12p70 from human monocytes and bone marrow derived dendritic cells, and selectively induces IL-12p40, an IL-12 antagonist [15-17]. There is also strong evidence for PGE2-mediated inhibition of the IL-12 receptor, diminishing IL-12 responsiveness [18]. Therefore the production of PGE2 in the environment of T cell activation has the potential to inhibit generation of a T helper type 1 mediated immune response. Additionally, resistance to re-infection requires the development of T helper type 1 immunological memory [19], and the IL-12 signaling pathway is necessary for an effector memory response [9]. Carrier state, relapse and re-infection may occur in both S. typhimurium and S. typhi related etiologies [20], with the implication that the initial infection does not bring about lasting immunity.
Studies in our laboratory have shown that macrophages and dendritic cells are activated by Salmonella to secrete a significant amount of PGE2 [21]. This response occurs both in vitro and in vivo, is COX-2 dependent, and is generated to a greater extent by viable pathogens than by killed bacteria or LPS [21]. This suggests that Salmonella stimulates production of conflicting mediators, and while this is a common theme in maintaining homeostasis, it may also be advantageous to the pathogen. Because the initiation of an adaptive immune response within this context may be hindered, it was of interest to determine the extent to which an antigen specific T helper type 1 response is inhibited by this particular feature of Salmonella infection. In studies presented here, DO11.10 TCR transgenic animals were used as a source of CD4+ T cells, and their response to antigenic stimulation in the presence of infected macrophages and dendritic cells was assessed. The relative contributions of COX-2, contact, and IL-12 were evaluated using this in vitro model.
MATERIALS AND METHODOLOGY
Isolation of Peritoneal Macrophages and Bone Marrow Derived Dendritic Cells
Peritoneal macrophages were isolated as previously described [22, 23]. Briefly, BALB/c mice were injected intraperitoneally with 250 μl of incomplete Freund’s adjuvant (Sigma Chemical Co., St Louis, MO). Four days later, the mice were euthanized, and peritoneal cavities were lavaged with RPMI-1640 (Gibco-BRL, Grand Island, NY) containing 2% FCS. Peritoneal cells were washed twice and then allowed to adhere to 12-well tissue culture plates (Costar, Cambridge, MA) for 45 minutes in RPMI-1640 containing 10% FCS before washing to remove non-adherent cells.
Bone marrow derived dendritic cells were isolated as previously described [24]. Briefly, femurs were flushed with RPMI-1640 containing 2% FCS to collect total bone marrow cells. Spicules and bone matrix were allowed to settle and removed. Total bone marrow cells were washed once and resuspended in RPMI-1640 containing 12% FCS and 1000 U/ml GM-CSF (PharMingen, San Diego, CA). Cells were fed every three days by adding 50% fresh medium. After 7 days in culture, non-adherent cells were removed, washed, and aliquoted into tissue culture plates in RPMI-1640 10% FCS without antibiotics, and exposed to medium or wild type Salmonella as described below.
All experiments using these animals were conducted following the guidelines set forth by the University of North Carolina at Charlotte Institutional Animal Care and Use Committee.
In Vitro Exposure of Macrophages or Dendritic Cells to Salmonella
Macrophages or dendritic cells cultured in RPMI-1640 10% FCS without antibiotics were briefly exposed to live Salmonella at ratios of 3:1 or 10:1 bacteria to cells. After 45 minutes, extracellular bacteria were removed by washing the cells and by the addition of gentamicin-containing medium. Culture supernatants were isolated from the cultures at the indicated times post-infection for PGE2, IFN-γ, or IL-4 analyses, respectively. In addition, some cells were treated with 1μM celecoxib (Pharmacia Corporation, Peapack, NJ) for 20 minutes prior to the addition of bacteria.
Stimulation of CD4+ T Cells
Spleens were harvested from euthanized BALB/c or DO11.10 TCR transgenic mice (The Jackson Laboratory, Bar Harbor, ME). Single cell suspensions were prepared by passage through a stainless steel sieve followed by lysis of red blood cells. CD4+ T cells were isolated with anti-mouse CD4 (L3T4) conjugated beads according to manufacturers’ instructions (Miltenyi, Auburn, CA). B lymphocytes were isolated from BALB/c splenocyte suspensions with anti-B220 conjugated beads (Miltenyi). DO11.10 CD4+ cells were cultured with equal numbers of BALB/c B cells as antigen presenting cells and stimulated with 1 μg/ml OVA peptide (OVA323-339) (Research Genetics, Huntsville, AL). CD4+ T cells from BALB/c animals were stimulated with 50 pg/ml recombinant IL-12p70 (PharMingen). Transwell cultures were performed in 12-well plates using 12 mm diameter polycarbonate membranes with 0.4 μm pores (Costar, Corning, NY). Anti IL-12 or isotype control antibodies (PharMingen) were used at a final concentration of 2 μg/ml.
Quantitation of PGE2 in Culture Supernatants
PGE2 levels in culture supernatants were analyzed using the Prostaglandin E2 Direct Biotrak Assay (Amersham Pharmacia Biotech, Piscataway, NJ), according to instructions from the manufacturer as previously described [21].
RESULTS
PGE2 Production is Stimulated by Infection of Elicited Peritoneal Macrophages with Salmonella Typhimurium
Previous studies in our laboratory have demonstrated that macrophages and dendritic cells respond to Salmonella infection with a robust inflammatory response, producing large amounts of PGE2 [21] This response has been shown to be dependent on COX-2 activity, since it is entirely abrogated by the introduction of the COX-2 specific inhibitor celecoxib. The amount of PGE2 secreted during an encounter with viable bacteria is far greater than that produced upon exposure to LPS or UV-killed Salmonella. These same cell types are responsible for PGE2 production in the mesenteric lymph nodes after oral inoculation with Salmonella [21]. Therefore PGE2 is present in the microenvironment of immune induction and may influence the initiation of an antigen-specific response. In order to examine the pattern of PGE2 production in the context of antigen-specific T cell priming during Salmonella infection, mixed cultures of DO11.10 CD4+ T cells, B cells, and elicited peritoneal macrophages or bone marrow-derived dendritic cells were prepared and stimulated with the Ova peptide antigen.
The results of one such experiment are shown in Fig. (1). Resting macrophages produced a low background level of PGE2, most likely as a result of constitutive expression of COX-1 (Fig. 1A). Culture supernatants from T and B cells contained only background levels of PGE2, even when antigen was added (Fig. 1A). When macrophages were infected with Salmonella typhimurium at a multiplicity of infection of either 3:1 or 10:1, PGE2 secretion increased by more than an order of magnitude (Fig. 1B). The addition of 1 μM celecoxib inhibited this response, indicating that PGE2 increases were due to COX-2 activity (Fig. 1B).
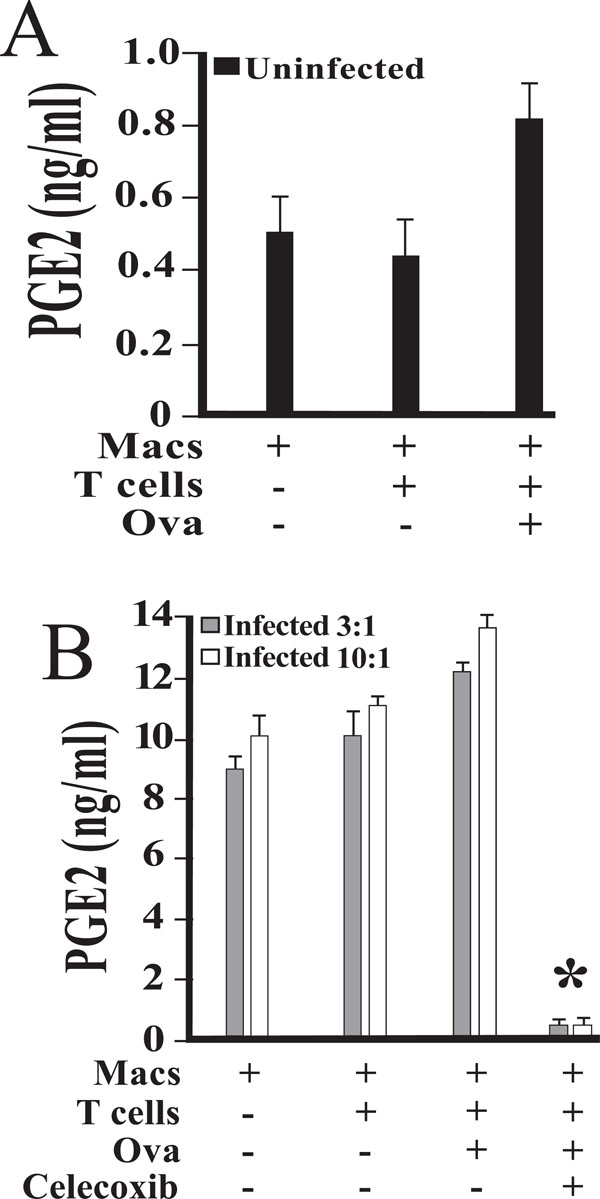
PGE2 production in mixed cultures of DO11.10 CD4+ T cells, B cells, and elicited peritoneal macrophages infected with Salmonella typhimurium. Peritoneal macrophages were isolated and cultured in the presence of medium alone (A) or viable Salmonella(B). The dose of bacteria initially added to each well was 3:1 (3) or 10:1 (10) bacteria to macrophage, as indicated. Following exposure for 45 minutes, cells were washed to remove any extracellular bacteria with gentamicin-containing medium. Magnetically separated splenic DO11.10 CD4 + T cells, B cells, and OVA peptide were added. Some cultures were supplemented with 1 uM celecoxib 20 minutes prior to infection. Culture supernatants were collected at 24 hours post-infection and stored at -80°C. Levels of PGE2 were measured by enzyme immunoassay, and are reported as mean values of triplicate determinations. An asterisk indicates a statistically significant difference (p < 0.01) when compared to cultures without celecoxib. These studies were performed three times with similar results.
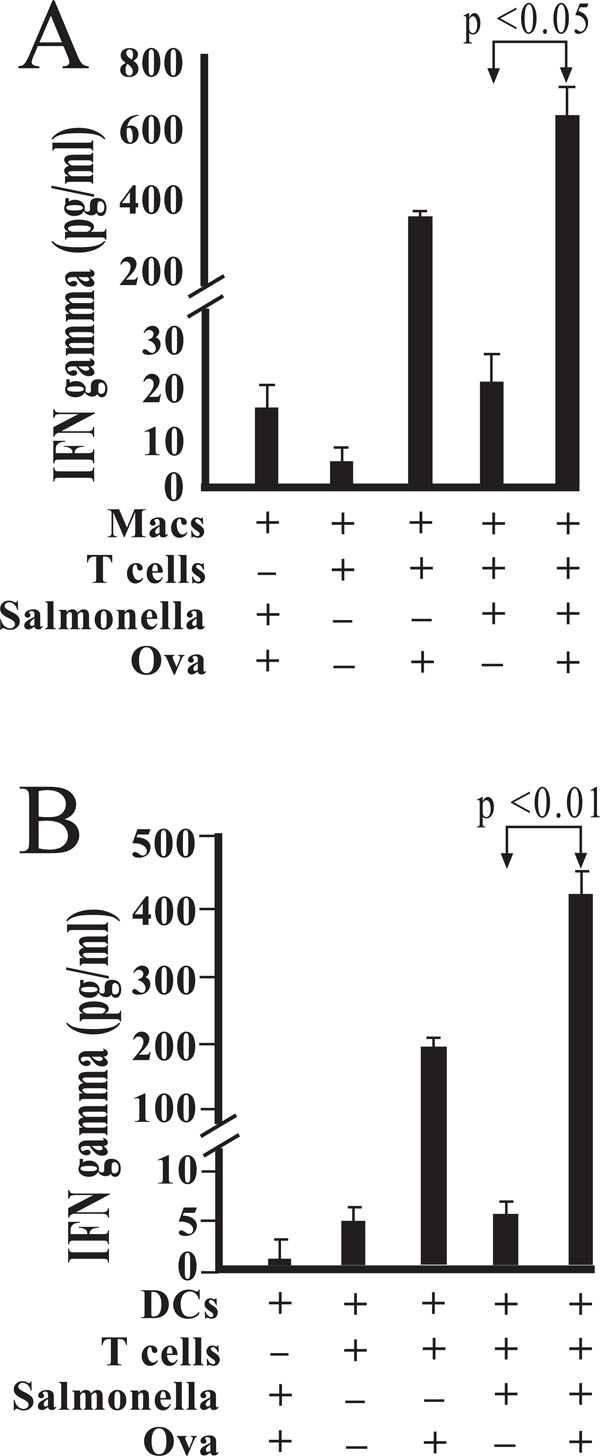
Salmonella-infected macrophages augment IFN-γ production by antigen-stimulated DO11.10 CD4+ T cells. Peritoneal macrophages were isolated and cultured in the presence of medium alone or viable Salmonella. Following exposure for 45 minutes, cells were washed to remove any extracellular bacteria with gentamicin-containing medium. Magnetically separated DO11.10 CD4 + T cells, B cells, and OVA peptide were added as indicated (A). In other studies, bone marrow-derived dendritic cells were infected with Salmonella and treated with gentamicin 45 minutes post-infection and prior to addition of DO11.10 CD4 + T cells, B cells, and OVA peptide (B). Levels of IFN-γ were measured by enzyme immunoassay, and are reported as mean values of triplicate determinations. These studies were performed three times with similar results.
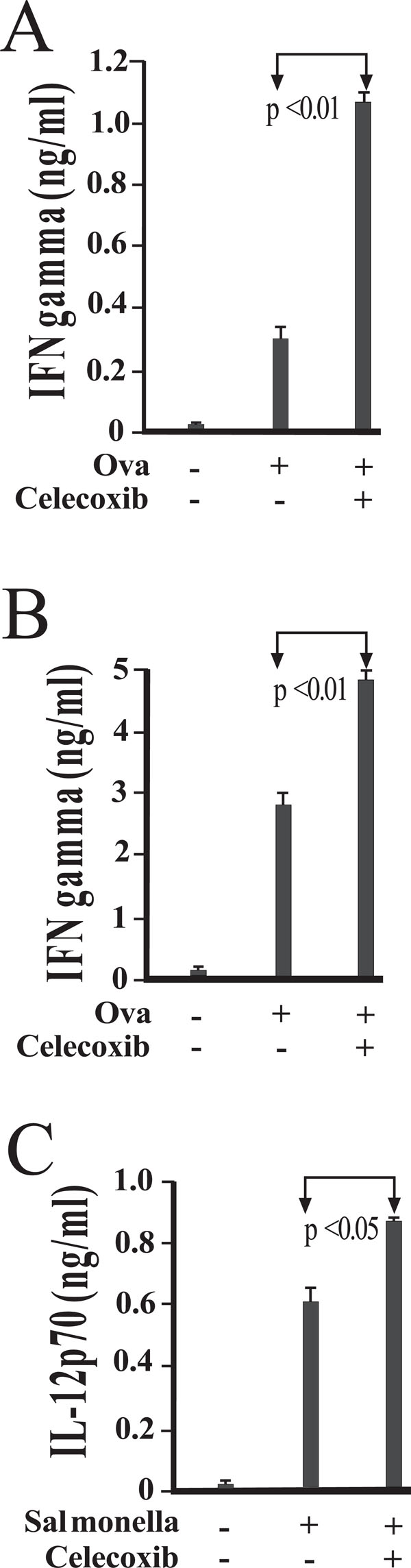
IFN-γ production in mixed and transwell cultures of DO11.10 CD4+ T cells and elicited peritoneal macrophages. T cells and macrophages were co-cultured or cultured in transwell. Peritoneal macrophages were isolated and cultured in the presence of medium alone or viable Salmonella. Following exposure for 45 minutes, cells were washed to remove any extracellular bacteria with gentamicin-containing medium. Magnetically separated splenic DO11.10 CD4 + T cells, B cells, and OVA peptide were co-cultured or separated by transwells (A). To evaluate the role of IL-12, some mixed cultures were supplemented with anti-IL-12p40 antibody or isotype control antibody (B). Levels of IFN-γ in 48-hour supernatants were measured by enzyme immunoassay, and are reported as mean values of triplicate determinations. These studies were performed three times with similar results.
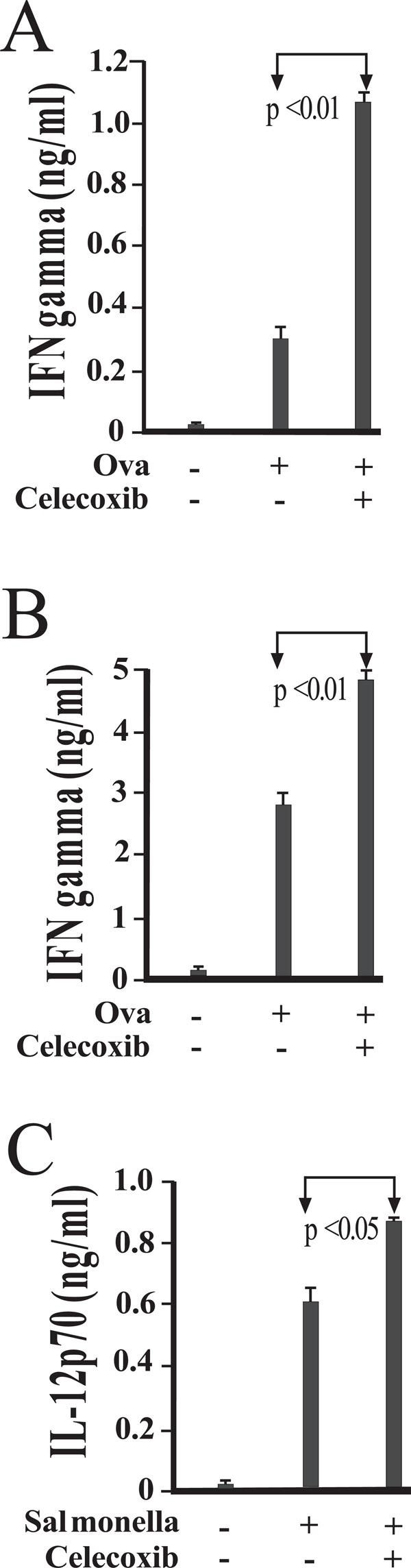
COX-2 activity influences IFN-γ and IL-12p70 production in antigen stimulated cultures of DO11.10 CD4+ T cells. Peritoneal macrophages were isolated and cultured in the presence of medium alone (A) or viable Salmonella(B). Following exposure for 45 minutes, cells were washed to remove any extracellular bacteria with gentamicin-containing medium. Magnetically separated splenic DO11.10 CD4 + T cells, B cells, and OVA peptide were added. To assess the role of COX-2, some cultures were supplemented with 1 uM celecoxib 20 minutes prior to infection. Levels of IFN-γ (A and B) and IL-12p70 (C) in 48-hour supernatants were measured by enzyme immunoassay, and are reported as mean values of triplicate determinations. These studies were performed three times with similar results.
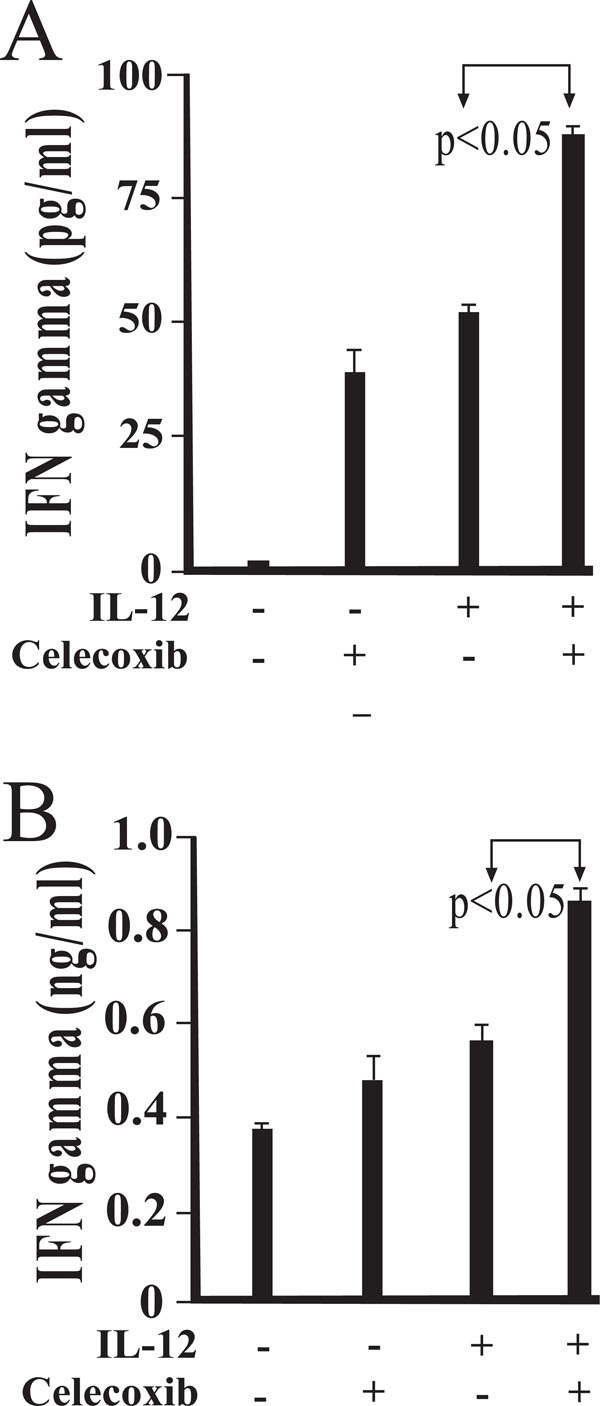
COX-2 activity influences IFN-γ production in IL-12 stimulated cultures of BALB/c CD4+ T cells. IFN-γ production by CD4 + T cells in the presence or absence of celecoxib was evaluated. Peritoneal macrophages were isolated and cultured in the presence of medium alone (A) or viable Salmonella(B). Following exposure for 45 minutes, cells were washed to remove any extracellular bacteria with gentamicin-containing medium. Magnetically separated splenic BALB/c CD4 + T cells were added and cultured in the presence of IL-12 and/or celecoxib. Results are presented as mean values of triplicate determinations. These studies were performed twice with similar results.
The same experiments performed with dendritic cells yielded similar results. Resting dendritic cells produced 0.26 ng/ml PGE2, versus 2.07 ng/ml when infected with Salmonella at a ratio of 3:1. The addition of T and B cells raised levels to 0.40 ng/ml in uninfected cultures with no further increase upon addition of the Ova peptide. Infected cultures produced 3.0 ng/ml PGE2 when T and B cells were present, and levels increased significantly with addition of antigen to 8.47 ng/ml (p=0.0002).
Antigen-Driven IFN-γ Production by CD4+ T Cells is Stimulated by Infection of Cocultured Peritoneal Macrophages with Salmonella Typhimurium
IFN-γ production via Salmonella-infected macrophages was exhibited by mixed cultures of DO11.10 CD4+ T and B cells (Fig. 2A). IFN-γ levels were significantly elevated (p < 0.05) by the combination of both antigen and the presence of infected macrophages when compared to similar mixed cultures in the absence of antigen.
When the corresponding studies were performed using bone marrow derived dendritic cells, a similar pattern of IFN-γ secretion was observed (Fig. 2B). Among Ova stimulated cultures, the amount of secreted IFN-γ was found to be significantly higher where dendritic cells had been exposed to Salmonella (p < 0.01). These results indicate that Salmonella-infected dendritic cells, like macrophages, were able to augment IFN-γ production in the presence of antigen (Fig. 2B).
Contact Inhibition by Transwell Coculture and Anti-IL-12 Each Partially Reduce the IFN-γ Response
In order to assess the individual contributions of contact and soluble factors to IFN-γ production, additional studies were performed with macrophages and DO11.10 CD4+ T and B cells. In Fig. (3A), macrophages were separated from T and B cells by transwell barriers. Cultures were stimulated with Ova peptide for 48 hours, at which time supernatants were collected and assayed for levels of IFN-γ. Prevention of physical contact between macrophages and T cells reduced the amount of IFN-γ produced by less than half, indicating that while some of this response is contact dependent, a significant portion is reliant on soluble factors (Fig. 3A).
To determine the nature of this soluble stimulation, anti-IL-12 antibodies were added to cultures (Fig. 3B). While IFN-γ levels were unperturbed by addition of isotype control antibody, reductions were observed in those cultures treated with anti-IL-12 (Fig. 3B). Taken together, the studies presented demonstrate the relative contributions of both cellular contact and IL-12 in the activation of antigen-specific IFN-γ production.
COX-2 Activity Influences IFN-γ Production in Antigen Stimulated Cultures of DO11.10 CD4+ T Cells
We next questioned whether Celecoxib could inhibit IFN-γ production in antigen stimulated cultures of CD4+ T cells. To address this question, DO11.10 CD4+ T cells and B cells, stimulated with Ova peptide, were incubated with untreated (Fig. 4A) or cultured with Salmonella-infected macrophages (Fig. 4B). In addition, some cultures were treated with 1 uM celecoxib to hinder COX-2 activity. IFN-γ levels are augmented by the addition of celecoxib in both uninfected (Fig 4A) and infected (Fig. 4B) co-cultures.
Analysis of supernatants from mixed cultures of macrophages and DO11.10 CD4+ T and B cells revealed that infection with Salmonella does elicit in vitro production of IL-12p70 (Fig. 4C). Inhibition of COX-2 with celecoxib treatment further increased IL-12 secretion, indicating that the endogenous PGE2 synthesis resulting from infection can reduce the IL-12 response (Fig. 4C).
COX-2 Activity Influences IFN-γ Production in Cultures Exposed to Recombinant IL-12
We next questioned whether treatment with celecoxib could alter IL-12-induced IFN-γ secretion in co-cultures of CD4+ T cells and macrophages. Fig. (5A) demonstrates that IL-12-induced IFN-γ production was significantly increased in the presence of celecoxib. More importantly, in Salmonella infected cultures, a significant increase in IFN-γ production was also observed with celecoxib treatment. Collectively, these results support the notion that COX-2 activity limits IL-12 induced IFN- γ production.
DISCUSSION
We have previously demonstrated a surprising potential for induction of PGE2 synthesis upon infection with Salmonella typhimurium both in cultured immune cells and lymphoid tissue [21]. High levels of PGE2 can be inhibitory for T cell-mediated immunity, a vital component of the host defense against Salmonella. The fact that PGE2 production occurs in the mesenteric lymph nodes demonstrates the presence of increased prostaglandins at mucosal sites following infection, and suggests that these mediators are present to influence the developing host response [21].
In the current study we began to address the impact of COX-2 activation and PGE2 production on IFN- γ production using co-cultures of antigen-specific CD4+ T lymphocytes and Salmonella-infected macrophages and dendritic cells. When DO11.10 CD4+ T cells are co-cultured with Salmonella-infected macrophages, they are exposed to elevated amounts of PGE2 (Fig. 1B). This in vitro result seems to mimic what occurs in vivo [21]. Salmonella-infected macrophages (Fig. 2A), or dendritic cells (Fig. 2B), augment the production of IFN-γ by DO11.10 CD4+ T cells in the presence of antigen. This antigen-stimulated IFN-γ production in macrophage-infected cultures is enhanced by cell/cell contact (Fig. 3A) or by IL-12 produced in these co-cultures (Fig. 3B). The presence of celecoxib significantly elevates the levels of IFN-γ (Figs. 4A and 4B) and IL-12p70 (Fig. 4C) produced in these antigen-stimulated co-cultures. Taken together these in vitro studies demonstrate that blocking COX-2 activity can significantly increase mediators (i.e. IFN- γ and IL-12) which augment T helper type 1 responses [3-7].
If these in vitro studies accurately depict what occurs in vivo following Salmonella infection, then they may have important implications for this bacterial infection being able to limit a protective T helper type 1 response. The ability of Salmonella infection to induce the production of PGE2in vivo [21] and in vitro (Fig. 1) likely serves to limit a T helper type 1 response [14-18]. While this response could be beneficial to the host by limiting a destructive, pro-inflammatory response, it may also limit the T helper type 1 response below what would be optimal for the immune response against this intracellular pathogen. Evidence to support this possibility comes from our previous work demonstrating that therapeutic administration of IL-12 can augment the protective host response against Salmonella [13]. The implication of this finding is that the host does not produce sufficient amounts of this cytokine to initiate a protective T helper type 1 response. The present work suggests that one mechanism which may be responsible for limiting IL-12 initiated IFN-γ is the presence of PGE2 induced by Salmonella. Future studies will be necessary to determine if treatment with COX-2 inhibitors can augment the protective T helper type 1 response against Salmonella in animal models.

