All published articles of this journal are available on ScienceDirect.

Expression and Characterization of a Small, Xylan/Cellulose-degrading GH43 Protein Derived from Biofertilizer Metagenome
Abstract
Background:
A putative glycosyl hydrolase gene biof1_09 was identified from a metagenomic fosmid library of local biofertilizers in previous report [1]. The gene is renamed as gh43kk in this study.
Methods:
The gene gh43kk, encoding a putative β-D-xylosidase was amplified by polymerase chain reaction (PCR) and successfully cloned and expressed in Escherichia coli. The expressed recombinant protein was purified by metal affinity chromatography. Its properties were initially verified by enzyme assay and thin layer chromatography (TLC).
Results:
The purified recombinant protein showed the highest catalytic activities at acidic pH 4 and 50°C toward beechwood xylan, followed by carboxymethylcellulose (CMC). TLC analysis indicated a release of xylose and glucose when xylan and CMC were treated with Gh43kk protein, respectively, whereas glucose and cellobiose were detected when avicel, cellulose and filter paper were used as substrates, suggesting its dual function as xylanase with cellulase activity. The enzyme indicated great stability in a temperature between 10 to 50 °C and a wide range of pH from 4 to 8. Enzyme activity of Gh43kk was enhanced in the presence of magnesium and manganese ions, while calcium ions, Ethylenediaminetetraacetic acid (EDTA) and sodium dodecyl sulfate (SDS) inhibited the enzyme activity.
Conclusion:
These results suggest that Gh43kk could be a potential candidate for application in various bioconversion processes.
1. INTRODUCTION
Lignocellulosic material is one of the potential sources for biomass energy production. It is the main basic component found in all plant cell walls and is mostly composed of three polymers: cellulose, hemicellulose and lignin, together with small amounts of other components, like acetyl groups, minerals and phenolic substituents [2]. Cellulose is a linear, unbranched homopolysaccharide of 10,000 to 15,000 D-glucose units connected by (β1→4) glycosidic linkage [3]. Hemicelluloses are heterogeneous polymers of pentoses (xylose and L-arabinose), hexoses (mannose, glucose and galactose), and sugar acids [2]. Lignin mostly consists of three different monomers, p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) arranged in branches with no constant organization [4].
In order to effectively utilize lignocellulosic resources as a substrate, cellulose and hemicellulose have to be broken down via hydrolysis to release sugar monomers, such as glucose and xylose. The conversion of lignocellulose to sugars could be accomplished by physical, chemical or enzymatic hydrolysis. However, high temperature and strong acids are mostly involved in the physical and chemical hydrolysis approaches, by which expensive, corrosion-resistant equipment are needed. Compared with those mentioned methods, the cost of enzymatic hydrolysis is rather low compared to acid or alkaline hydrolysis because enzyme hydrolysis is usually conducted at mild conditions (pH 4.8 and temperature 45–50°C) and does not have a corrosion problem [5, 6].
Cellulolytic enzymes found in nature are produced by bacteria, fungi, protozoans, plants and animals. However, the majority of environmental microorganisms, with an estimation of about 85-99%, cannot be cultured by conventional laboratory cultivation methods [7]. In order to avoid the limitation of culturing method, the metagenomic approach has been introduced. Metagenomics is the genomic analysis of microorganisms by direct extraction and cloning of environmental DNA [8]. This method leads to the discovery of many novel cellulolytic enzymes such as RmCel12A glycoside hydrolase family 12 cellulase (LC-CelA) [9] and Cel5R, a novel halostable endoglucanase obtained from soil metagenomic library [10], Zfyn184 a cellulase/ hemicellulase belonging to glycoside hydrolase family 44 [11].
In our previous work, we reported a clone from metagenomics library constructs showing activities toward both cellulose and xylan [1]. After sequencing the positive library construct, an open reading frame of a glycosyl hydrolase gene was predicted. The amino acid sequence of the translated nucleotides shared similarity with the conserved domain of glycosyl hydrolase family 43 (GH43) [1]. In this work, the above mentioned open reading frame, renamed as gh43kk gene, was cloned, expressed, and the encoded protein was purified, and its biochemical properties were characterized.
2. MATERIALS AND METHODS
2.1. Cloning of gh43kk Gene
The gh43kk gene was obtained from a nucleotide sequence of a metagenomic subclone Biof1_09 (Genbank JQ581599 [1]. The gene was amplified by polymerase chain reaction (PCR) using specific primers containing BamHI and EcoRI restriction site (5’-GGGCGCGGATCCATGTACGCCAT GAAAAA CATTTTTAAATATG-3’) and (5’-GGGCCGCCGGAATTC TGTAGAAGTGCCACCTTTCAAC -3’). The amplification protocol was according to the Taq DNA polymerase manual (Thermo Scienctific, USA), starting with an initial denaturation at 95°C for 3 min, 30 cycles of denaturation at 95°C for 30 s, annealing at 42 °C for 1 min 30 s and extension at 72°C for 1 min 30 s, final extension at 72°C for 10 min. The PCR product was purified by QIAquick® PCR Purification Kit (Qiagen, Germany) and was digested with FastDigest BamHI and EcoRI (Thermo Scienctific, USA). Afterward, the gh43kk fragment was inserted into pET28a(+) expression vector (Novagen, USA) by T4 DNA ligase (New England Biolabs, USA). The recombinant expression vector (pET/gh43kk) was transformed into E. coli BL21 by heat-shock technique. The transformants were selected by colony PCR and then also confirmed by sequencing (Macrogen, Korea).
2.2. Bioinformatics Analysis and Three-dimensional Structure Modeling
The molecular weight and isoelectric point of the translated nucleotides of the gene gh43kk were calculated by Expasy software (https://web.expasy.org/compute.pi/). The patterns of amino acid residue conservation and divergence in a protein family were analyzed by searching in the Conserved Domain Database (CDD, https://www.ncbi.nlm.nih.gov/Structure/cdd /cdd.shtml).
A three-dimensional model of Gh43kk was generated by RaptorX (http://raptorx.uchicago.edu/StructurePrediction/ myjobs/62894490_578596/) with a score of 32. The P-value for the relative global quality was 0.0365. The GDT (global distance test) and uGDT (un-normalized GDT) values of the model were 30 and 33, respectively.
2.3. Expression and Purification of Gh43kk Protein
The expression of the recombinant protein was done under low temperature as follows: OD600 of overnight bacterial culture was adjusted to 0.2 and the incubation was continued at 37°C until the OD600 reached 0.4. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.1 mM, and the cell suspension was further incubated for 20 h at 16°C. Lysates were prepared from 100 ml E. coli culture. Bacterial cells were harvested by centrifugation at the speed of 7,500 rpm, 4 °C for 10 min. Each gram of the resulting pellet was lysed in 5 ml of 50 mM sodium acetate pH 5, 300 mM NaCl, 20 mM imidazole and 90% (v/v) B-PER™ Bacterial Protein Extraction Reagent (Thermo Scientific, USA). Lysozyme (final concentration 1 mg/ml (w/v) and cOmplete™ Protease Inhibitor Cocktail (Sigma-Aldrich, USA) was further added to the lysate, then incubated at room temperature for 30 min prior to sonication. Sonication (Vibra-Cell™, Sonics, was done at 35% amplitude (pulse duration of 5 s on and 10 s off) on ice. The lysate was centrifuged at 18,000 g for 30 min at 4°C and the supernatant was sterile filtered through 0.22 µm membrane filter. Protino® Ni-NTA Agarose (Macherey Nagel, Germany) was used for purification according to the manufacturer's instruction. Briefly, 5 ml of HisPur™ Ni-NTA Resin (Thermo Fisher Scientific, USA) was loaded into an empty gravity flow column with filter frits (Protino® column 14 ml, Macherey-Nagel, Germany). The agarose was equilibrated with lysis buffer. The clear lysate was then added into the column, and incubated with the Ni-NTA agarose with gentle shaking on an orbital shaker for 60 min at 4°C. After letting the lysate flow through the column, the agarose was washed twice with 50 mM sodium acetate pH 5, 300 mM NaCl, and 30 mM imidazole. The recombinant protein was eluted with 50 mM sodium acetate pH 5, 300 mM NaCl, and 300 mM imidazole. The eluate was then brought through the Vivaspin® 6 (cut off 10 kDa; GE Healthcare) to change the buffer to 20 mM Sodium acetate buffer pH 5.4. The purified protein was checked by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using 13.5% acrylamide-separating gel. Western blot analysis using anti-his tag monoclonal antibody was done to confirm the recombinant protein. Concentration and yields of the purified protein were calculated by Bradford protein assay. Purity and recovery of the recombinant proteins were estimated from protein band intensity in SDS-PAGE quantified by ImageJ software (National Institutes of Health, USA).
2.4. Agar Well Diffusion Test
Azurine cross-linked cellulose (AZCL-HE-cellulose) (Megazyme, Ireland) or beechwood xylan (Megazyme, Ireland) was mixed with molten sterile agar (prepared with 50 mM sodium acetate buffer pH 5) to a final concentration of 1% (w/v). Various amounts of the purified protein, as well as the commercial cellulose mixture, Celluclast™ (Novozymes, Denmark) and Cellubrix® (Novozymes, Denmark) as positive control and protein elution buffer (0.1 M sodium acetate buffer pH 5) containing imidazole as negative control were dropped into agar wells. After incubation at 37°C for 2 days, the plate containing beechwood xylan was stained with 0.33% Gram’s iodine and clear zones around the agar wells were observed. When the plate containing AZCL-HE-cellulose was used, smooth blue zones around the agar well indicated the activity of cellulose-degrading enzymes.
2.5. Enzyme Activity Assay and Kinetics
Enzyme activity was determined by incubating substrates with the purified enzyme in 100 mM sodium acetate buffer pH 5 at 50°C for 30 min, which will be used as a standard condition for the rest of the enzyme activity experiment. Reducing sugars were assayed by the DNS method with glucose as a standard. The concentration of pulverized substrates (avicel, beechwood xylan, cellobiose, cellulose, carboxymethyl cellulose) was 1% (w/v), while the size of filter paper was 0.5×1.5 cm. Negative controls were done by incubating the substrates without the enzyme (using elution buffer instead). The values of colorimetric detection (in reducing sugars) of controls were subtracted from the results of the actual reactions. All the experiments were done in three replicates. One unit of enzyme activity was defined as the amount of enzyme that releases 1 µmol of glucose equivalents per minute. Specific activity was exhibited in the unit of activity enzyme in one mg of protein. Protein concentration was determined by using the method as described by Bradford [12] with BSA as a standard.
The kinetic studies were determined using beechwood xylan concentrations from 0.5 to 5 mM, respectively. Reducing sugars were measured after the reactions were incubated at 50°C for 30 min. The Michaelis constant (Km) and maximum velocity (Vmax) were determined by means of Lineweaver–Burk plots [13]
2.6. Thin Layer Chromatography (TLC)
TLC was performed as modified by Zhou and coworkers [14]. One percent (w/v) of substrates were incubated with Gh43kk protein under optimal conditions. Subsequently, aliquots of 2 μl each were spotted on thin layer chromatography (TLC) silica gel 60 plates (Merck, USA), along with standards of glucose, cellobiose, xylose, and arabinose, respectively. Separation was carried out by TLC with a mobile phase (ethanol/1-butanol/water at a ratio of 3:5:2) at room temperature. Sugars were detected by spraying the plates with ethanol and sulfuric acid (ratio 95%:5% v/v), followed by heating at 100°C for 10 min. The experiment was done in triplicates. Relative rates of flow (Rf) were calculated as Rf = X/ Y, by which X = migration distance of the sample; and Y = migration distance of the solvent.
2.7. Effect of Temperature, pH, Metal Ions and other chemicals on the Recombinant Enzyme
The optimal temperature for enzyme activity was performed at various temperatures ranging from 10 to 90°C. To determine temperature stability of the enzyme, the purified enzyme was incubated at different temperatures for 30 min prior to the enzyme activity assay. Determination of optimum pH was done in 100 mM sodium acetate buffer (pH 3-6), 100 mM phosphate buffer (pH 6-8) and 100 mM tris-HCl buffer (pH 8-10). For the test of pH stability, the protein was pre-incubated in buffers with different pH for 1 h at 4°C, then the activity was assayed under the same condition as mentioned in Enzyme activity assay section. The effects of metal ions compounds (Ba2+ Ca2+ Co2+Cu2+, Fe2+, Li+, Mg2+, Mn2+, Ni2+, Sr2+ and Zn2+, salts (KCl, NaCl), chelating agent (Ethylenediaminetetraacetic acid; EDTA), and 1% (w/v) detergent (sodium dodecyl sulfate; SDS) were determined under standard condition. The concentrations of metal ions, salts and EDTA were 5 mM. Control was buffer without addition of any tested chemicals or metal ions. Beechwood xylan was used as substrate for all experiments.
3. RESULTS
3.1. Bioinformatic Analysis and Modular Structure of the Gh43kk Protein
The gene gh43kk contains putative nucleotides of 330 bp, which encode a polypeptide of 110 amino acid residues with a theoretical molecular mass of 12.81 kDa and an isoelectric point of 4.5. The protein Gh43kk shared conserved regions within the amino acid sequences of the glycosyl hydrolase family 43. Among those are amino acid residues which were reported to be found on the active site of the enzyme, namely aspartic acid (Asp43), glutamine (Gln93), glycine (Gly107), and Threonine (Thr108) (Graciano 2012) The multiple amino acid sequence alignment in Fig. (1) shows that these key residues are well conserved in Gh43kk protein.
As shown in the three-dimensional model (Fig. 2), the N-terminal region of Gh43kk displays a β-meander motif containing 4 consecutive antiparallel β-strands linked together by hairpin loops. The C-terminal region shows a v-shaped helix-break-helix fold [15]. The conserved catalytic aspartate (Asp43) is located in the hairpin loops linking the first pair of the antiparallel β-sheets.

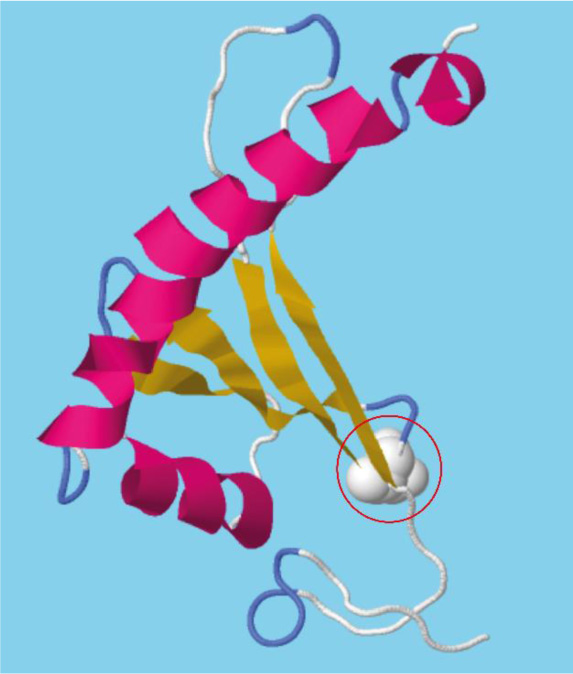
3.2. Cloning, Expression and Purification of the Recombinant Gh43kk Protein
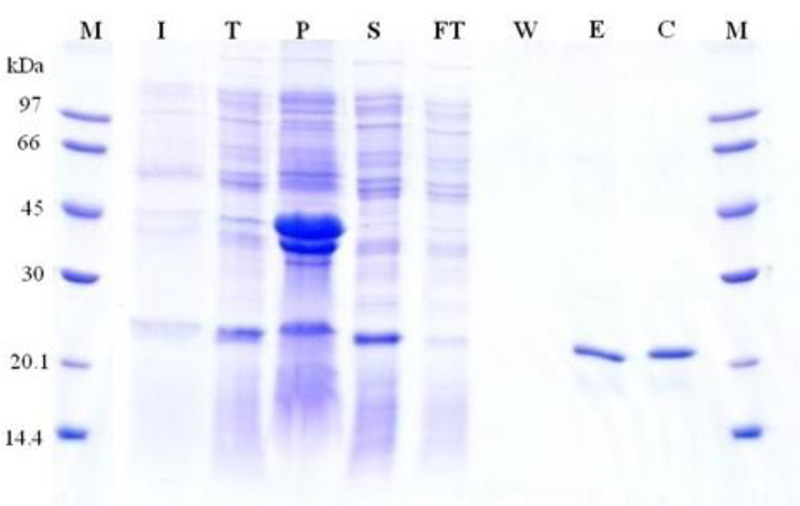
The gene gh43kk was successfully cloned, and protein expression was optimized. The recombinant Gh43kk protein was effectively overexpressed at 37°C by using IPTG as an inducer, however, a large amount of the expressed protein remained as an inclusion body (Fig. S1), which obstructs the purification. The recombinant protein was soluble when the temperature was shifted to 16°C, therefore, the recombinant protein Gh43kk was successfully expressed and purified at that temperature (Fig. 3).
Fig. (3) shows the purification step with the purified protein band of about 23 kDa. The purified recombinant protein was more than 95% pure, with around 75% recovery. Approximately 10 milligrams of recombinant protein was obtained from 1 liter of bacterial expression culture (1-1.5 g wet weight). The recombinant Gh43kk was confirmed by Western blot analysis and a protein band of about 23 kDa was visualized (Fig. 3B).
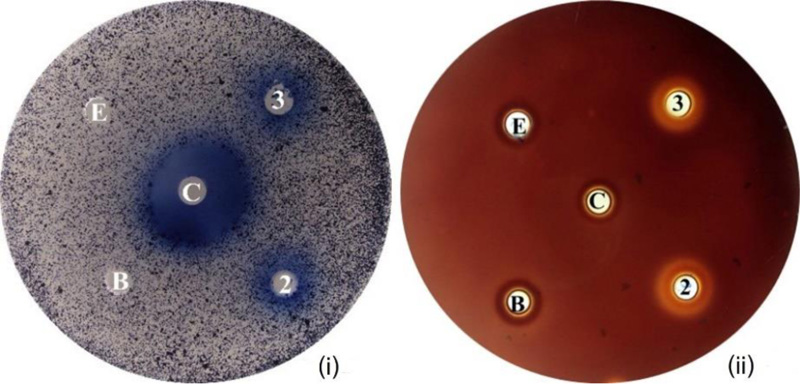
3.3. Substrate Specificity of the Purified Recombinant Protein Gh43kk
Activities of the purified Gh43kk protein were detected both on agar and in liquid suspension using various substrates. Smooth blue-colored zone or clear zones were observed on agar plates containing AZCL-Cellulose and xylan as substrates, respectively (Fig. 4). The same result was observed by spectrophotometric experiments, by which the enzyme showed the highest hydrolytic activity towards the beechwood xylan with a specific activity of 286.37 ± 7.37 mU·mg-1 protein followed by CMC with 129.78 ± 7.91 mU·mg-1 protein, respectively (Table 1). The Km for Gh43kk on beechwood xylan was 0.153 µM, while the Turnover number (Kcat) was 0.0213, and Kcat/Km of the enzyme was 0.143.
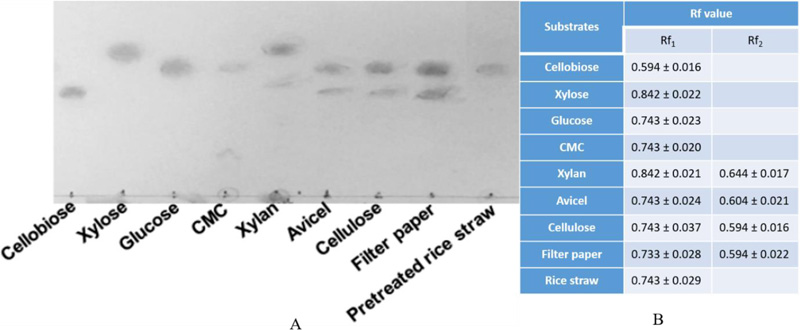
Thin Layer Chromatography (TLC) analysis of hydrolysate of different substrates treated by the recombinant protein Gh43kk revealed several patterns of sugar products (Fig. 5). Both glucose and cellobiose were detected when avicel, cellulose and filter paper were hydrolyzed with Gh43KK. Only glucose was observed when CMC and rice straw were degraded with the recombinant enzyme. Gh43KK also exhibits its xylanase activity since xylan could be hydrolyzed as well, yielding xylose as a product.
3.4. Effect of Temperature, pH, Metal Ion, and other Chemicals on Activity and Stability of the Recombinant Protein Gh43kk
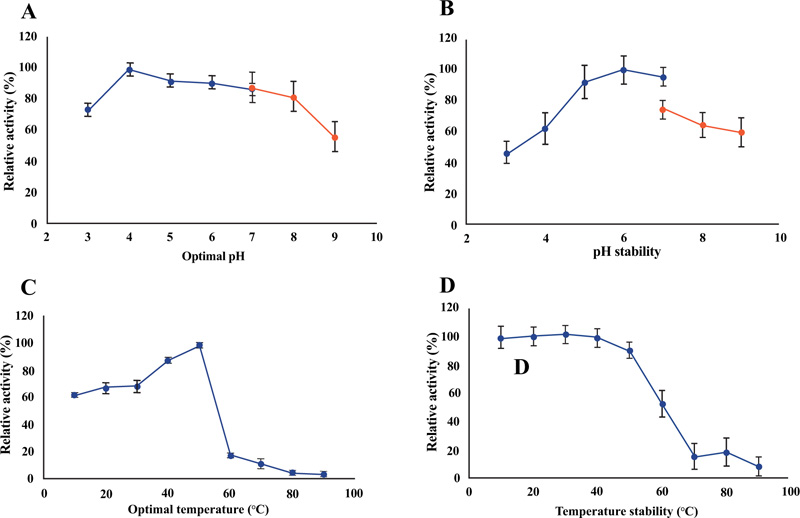
The purified recombinant Gh43kk could work well (with an activity of more than 80%) in pH ranging from 4 to 8, with an optimum pH of 4 (Fig. 6A). However, the enzyme activity of about 60% was still observed at pH 3 and 9, suggesting its capability in working at a very wide range of pH (Fig. 6A). The enzyme maintained its activity of more than 60% when pre-incubated overnight in buffer with pH ranged from 4 to 9, where pH 5 to 7 was shown to have the least effect on its activity (Fig. 6B). The recombinant protein Gh43kk showed increased activity when the temperature rose toward 50°C, which was also the optimum temperature for the enzyme (Fig. 6C). Its activity decreased rapidly when the temperature exceeded 50°C (Fig. 6C). The recombinant protein maintained more than 80% of its activity after pre-incubation at the temperature under 50°C (Fig. 6D).
The highest activity (with an activity of about 120%) of the recombinant enzyme was observed when manganese ions were present. Slightly higher activity of the enzyme was also observed with the presence of magnesium ion, however it did not significantly enhance the activity of the enzyme (p-value of 0.21). Moderate inhibition (activity of about 76-79%) of the enzyme Gh43kk was observed when copper, barium, potassium and sodium ions were present. Metal ions of zinc, calcium and strontium, and sodium dodecyl sulfate (SDS) were shown to strongly inhibit the activity of the recombinant enzyme. Gh43kk protein almost lost its activity when incubated with Ethylenediaminetetraacetic acid (EDTA) (with an activity of about 4.68%) (Table 2).
Table 1.
| Substrate | Specific Activity (mU-mg-1) | Relative Activity (%) |
|---|---|---|
| Avicel | 0.73± 0.20 | 0.25 |
| Beechwood xylan | 286.37± 7.37 | 100.00 |
| CMC | 129.78 ± 7.91 | 45.00 |
| Cellobiose | 17.37± 1.86 | 6.07 |
| Cellulose | 3.80± 0.94 | 1.33 |
| Filter paper | 0.31± 0.08 | 0.11 |
| Agent | Relative Activitya (%) | Agent | Relative Activity a (%) |
|---|---|---|---|
| None* | 100± 6.48 | Mn2+ | 120.52 ± 6.91 |
| Ba2+ | 76.12 ± 5.33 | Na+ | 79.25 ± 3.19 |
| Ca2+ | 38.07 ± 3.80 | Ni2+ | 48.57 ± 3.49 |
| Co2+ | 64.97 ± 3.56 | Sr2+ | 25.16 ± 2.61 |
| Cu2+ | 61.04 ± 9.32 | Zn2+ | 42.86 ± 7.71 |
| Fe2+ | 96.26 ± 3.56 | EDTA | 4.68 ± 3.85 |
| K+ | 78.44 ± 3.06 | SDS | 34.22 ± 6.41 |
| Li+ | 59.97 ± 3.30 | TritonX-100 | 69.61 ± 2.04 |
| Mg2+ | 108.57 ± 6.98 | - | - |
4. DISCUSSION
Small proteins (SPs) are loosely defined as polypeptides that contain ≤100 amino acids, while some works suggested greater thresholds for SPs of ≤200 amino acids [16, 17]. In general, SPs contain at least a basic domain, which is sufficient to perform or assist a biological process. Previous studies suggested that the length of a protein sequence is connected to its specific functions and that SPs might show not many remarkable functions compared with large proteins [18]. However, there seems to be an important tendency in evolution favoring shorter rather than longer proteins for specific functions [19].
In this work, we demonstrated a small protein with a complete bifunctional activity as xylanase and cellulase despite its small size. As shown by the multiple alignments of the amino acid sequence of Gh43kk, the protein contains amino acid residues which were reported to be found on the active site of enzyme members in the glycosyl hydrolase family 43, namely Asp42, Gln93, Gly107, and Thr108. Some of these amino acid residues play critical roles in enzyme activity, such as Asp42, which was reported to play a major role in modulating pKa and in retaining the right position of the active site residues [20, 21].
A bifunctional enzyme is an enzyme that contains two different catalytic properties in the same polypeptide chain [22]. Utilization of both hexoses and pentoses sugars present in lignocellulosic biomass could be an important step to decrease production costs for many bioconversion processes. Therefore, a bifunctional xylanase/ cellulase is certainly more valuable than single xylanase or cellulase since it can degrade both the hemicellulose and cellulose fraction of the biomass simultaneously. Furthermore, the problem in the mashing process caused by viscous pentosans can be solved when both xylanase and cellulase are present in the fermentation mixture [23].
Several conserved amino acids indicating the predicted active site were found in the primary structure of the Gh43kk modules. This might indicate a minimal requirement for the catalytic activity of enzymes in this family. In general, GH43 enzymes exhibit a five-bladed β-propeller fold (clan F) [21]. The active site is found extending far down in the cavity at the core of the β-propeller. In our work, the β-meander motif found in the structural model of Gh43kk would possibly represent one single blade out of the five blades of the β-propeller module of GH43 enzymes [21]. One important conserved residue, an aspartate (Asp43) might play a crucial role as a catalytic acid for the hydrolysis activity of this small enzyme. This residue is found at the hairpin loop of the peripheral β-sheet. The position of the Asp43 residue at the hairpin loop seems to share a similar position of the catalytic aspartate (Asp46) at the center of the active site of the GH43 five-bladed β-propeller module [21]. Hence, this result might suggest the most functional domain which is sufficient for the catalytic ability of the GH43 enzyme. However, the activity of Gh43kk is low when compared to other enzymes in the same categories, as cited in Table 3. This might be due to its small size, which does not fulfill the complete configuration of the active site of the GH43 enzyme reported earlier. Nevertheless, this result might indicate a minimal domain/residue requirement for the functionality of the GH43 enzyme.
Inclusion bodies have been frequently reported when recombinant protein is expressed at a high level in Escherichia coli, which results in aggregation of the highly expressed protein molecules [24]. Causes of high expression of recombinant protein include using high temperature during protein expression, high concentration of inducer, and expression under strong promoter systems, which result in expression of the target protein at a high translational rate [25]. Expression of recombinant protein at low temperature has been considered to support the formation of “non-classical” inclusion bodies [26]. These non-classical inclusion bodies are soft, easily extractable by using non-denaturing solvents [27]. In general, overall cell processes slow down at low temperature, leading to a reduction rate of transcription, translation; hence recombinant proteins are produced in a lower amount. Furthermore, the degradation of proteolytically sensitive proteins also declines at low temperature. Lowering the concentration of the induction agent also improves the solubility of the recombinant protein by reducing the transcription rate as well.
The observed size of about 19 kDa of the recombinant protein in SDS-PAGE is slightly larger than the predicted size (12.81 kDa). These might be due to a decrease in protein mobility, which can be a result of post-translational modification in the possible SDS-binding sites, such as glycosylation, phosphorylation of serine, threonine and tyrosine, or sulfation of tyrosine [28]. Those alterations could change the internal hydrophobicity of the protein and hence slow down the protein migration observed in SDS-PAGE. Furthermore, the presence of additional chemicals in protein preparation, such as salts, detergents, or organic solutions, can result in the same consequence since they can alter or distort the protein or many of them might take part or obstruct the binding of SDS with the protein.
Substrate specificity of the recombinant enzyme Gh43kk was verified using three different methods, namely spectrophotometric analysis of released reducing sugar, agar well assay, and TLC analysis. The protein Gh43kk shows activities toward CMC and xylan, thereby confirming the previous finding about the dual activities of the Biof1_09 crude cell-free extract [1]. However, the treatment condition of rice straw has to be further investigated since xylose was not detected when rice straw was hydrolyzed under the given condition. This might be due to harsh treatment with acid and high temperature in the pretreatment condition that could degrade the loose hemicellulose structure (including xylose), thereby leaving only the rigid cellulose after pretreatment.
Gh43kk shows comparable properties with the Biof1_09 crude cell-free extract, including pH optimum (pH 4), optimal temperature (50°C), a wide range of pH stability (from pH 3–9) and stability at a temperature below 60°C, as reported earlier by the authors [1]. Some enzymatic properties of the recombinant Gh43kk protein were compared with other GH43 enzymes derived from metagenomics (Table 3). Most of them work effectively under relatively high temperatures (40-55°C) and near-neutral pH (pH 5.5-7). However, it is notable that Gh43kk has an optimal pH for its activity in acidic conditions (pH 4).
Differences in metal ion sensitivities among the GH43 enzymes have been reported earlier [29]. To determine further properties of the enzyme Gh43kk in this study, several chemicals and metal ions were applied with the enzyme and its activity was monitored. Similar observation was made with CoXyl43, a GH43 β-xylosidase/α-arabinofuranosidase, which was inactivated by zinc and copper ions but was activated by manganese ions [30]. Chelating reagents such as EDTA had a negative impact on Gh43kk activity, which leads us to suggest that Gh43kk might be a metFalloenzyme. However, other GH43 enzymes, such as HiAbf43 and XynAMG1, do not require metal ions for their activities [31].
| Functions | Source | Substrate | Protein (kDa) | Optimum Temp (°C) | Optimum pH |
Activity (U·mg-1) |
References |
|---|---|---|---|---|---|---|---|
| Xylanase/ cellulase | biofertilizer | Beechwood xylan | 23 | 50 | 4 | 0.29 | This study |
| β-xylosidase | soil | pNPX | 64.4 | 40 | 6 | 0.012 (12 mU·mg-1) |
[31] |
| β-xylosidase/α-arabinofuranosidase | compost | birchwood xylan | 36.19 | 55 | 7.5 | 22.0 | [32] |
| β-xylosidase | gut bacterial genome | pNPX | 61.07 | 50 | 6.5 | 2.83 ± 0.23 | [33] |
| endo- β -1,4-xylanase | Cow rumen | beechwood xylan | 99 | 50 | 6 | 709 | [34] |
| β -D-xylosidase/α-L-arabinofuranosidase | rumen | pNPX | 42 | 40 | 7 | 36.3 | [35] |
CONCLUSION
The enzyme in this study is one of the smallest GH43 enzymes ever reported so far. It displays a notable ability to work under acidic conditions, whereas other reported enzymes in the same family could work only at near-neutral pH. With its dual activities as xylanase and cellulase, and its ability to work under acidic condition and high temperature, Gh43kk enzyme could be an interesting candidate for large-scale expression for applications in industrial processes that require acidic conditions and high temperatures such as pulp and paper industries.
LIST OF ABBREVIATIONS
| Asp43 | = Aspartate |
| SPs | = Small Proteins |
| EDTA | = Ethylenediaminetetraacetic Acid |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available within the article.
FUNDING
This study was funded by the National Research Council of Thailand (Grant number 570017).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors thank Mr. Pumi Eawpadung for laboratory assistance, and Dr. Piyanun Harnpicharnchai for the critical discussions. We also thank Assoc. Prof.Dr. Soontorn Oraintara for assistance in statistical analysis. We are also grateful to the Protein and Proteomics Research Center for Commercial and Industrial Purposes (ProCCI), Khon Kaen University, Khon Kaen, Thailand, for additional laboratory support.

