All published articles of this journal are available on ScienceDirect.

Different Distribution of Core Microbiota in Upper Soil Layer in Two Places of North China Plain
Authors Info & Affiliations
Abstract
Backgrounds:
Soils harbor diverse bacteria, and these bacteria play important roles in soil nutrition cycling and carbon storage. Numerous investigations of soil microbiota had been performed, and the core microbiota in different soil or vegetation soil types had been described. The upper layer of soil, as a source of organic matter, is important and affected by the habitats and dominant bacteria. However, the complexity of soil environments and relatively limited information of many geographic areas had attracted great attention on comprehensive exploration of soil microbes in enormous types of soil.
Methods:
To reveal the core upper layer soil microbiota, soil samples from metropolis and countryside regions in the North China Plain were investigated using high-throughput sequencing strategy.
Results:
The results showed that the most dominant bacteria are Proteobacteria (38.34%), Actinobacteria (20.56%), and Acidobacteria (15.18%). At the genus-level, the most abundant known genera are Gaiella (3.66%), Sphingomonas (3.6%), Acidobacteria Gp6 (3.52%), and Nocardioides (2.1%). Moreover, several dominant operational taxanomy units OTUs, such as OTU_3 and OTU_17, were identified to be associated with the soil environment. Microbial distributions of the metropolis samples were different from the countryside samples, which may reflect the environments in the countryside were more diverse than in the metropolis. Microbial diversity and evenness were higher in the metropolis than in the countryside, which might due to the fact that human activity increased the microbial diversity in the metropolis.
Conclusion:
The upper layer soil core microbiota of the North China Plain were complex, and microbial distributions in these two places might be mainly affected by the human activity and environmental factors, not by the distance. Our data highlights the upper layer soil core microbiota in North China Plain, and provides insights for future soil microbial distribution studies in central China.
1. INTRODUCTION
The soils harbor abundant microbial resources and contain high numbers of microbes [1-3]. Among these microbes, bacteria play important roles in various aspects, especially in carbon storage and nutrient cycling, for example, they can promote the cycling of C, N, S, and P, which can help the plant grow [4-8]. Especially, the upper layer of the soil generally contains many organic minerals, which are mainly affected by habitats and soil microbiota [9]. Moreover, soil microbiota can help remove pollutants and provide most of the antibiotics in clinical use today [3, 10]. The environmental factors played important roles in microbial distribution, while the geographic distances showed little effect on microbial diversity in soil [11-14]. A global analysis of drylands indicates that increasing aridity reduces soil microbial diversity [11]. The microbial community during corpse decomposition in different vegetation soil types are similar, and the dominant factor driving microbiota development is the nitrogen and carbon input [15]. Moreover, deforestation would affect the soil microbiota and the alpha diversity would be increased after the slash-and-burn forest cleaning in Amazon [16].
The microbial distribution in different biogeographical areas is different, and the dominant bacteria in soils worldwide are Proteobacteria, Actinobacteria, Verrucomicrobia, Acidobacteria and Firmicutes [17, 18]. The most dominant bacteria in drylands are assigned to Actinobacteria, which composes 23%-29% of the total bacteria [11], and the desert soil microbiota is distinct from microbiotas of non-desert soils [19]. In relatively undisturbed soil samples collected from North America, the most dominant bacteria are Acidobacteria, Veruucomicrobia and Bacteroidetes [20, 21]. The investigation of the East European plain soils showed that the most abundant bacteria are Actinobacteria, Proteobacteria and Verrucomicrobia [22]. The soil microbial diversity is affected by vegetation type, and the rhizospheric microbial distribution of different plants is distinct [23, 24]. Despite many efforts that have been tried to understand global soil microbial distribution, such as the Earth Microbiota Project [25], the microbial distribution in many geographic areas is still unknown.
Here, we present an upper layer soil microbiota study to assess the microbial diversity using a high-throughput sequencing approach in two different areas in the North China Plain. The sampling places have been used for agriculture for thousands of years, encompassing the countryside and the metropolis. We analyzed the dominant microbes in these samples and compared their microbial distribution. Moreover, the potential relationship between the soil samples and the environmental factors is discussed.
2. MATERIALS AND METHODS
2.1. Sample Collection and Analysis
The 13 soil samples were collected from two different regions in the North China Plain, Xincai county and Zhengzhou of Henan province, China, in March 2018 (Table 1). Sampled soils are moist clay in these two places (Fig. S1). Among them, 7 soil samples were collected from Xincai county (countryside place, named as XC group), and another 6 samples were collected from Zhengzhou (metropolis, about 300 kilometres from Xincai, named as ZZ group). The soil samples were collected from 5-10 cm of the soils, and were transferred to the laboratory and stored at -20 °C before use (Table 1). To measure pH, 0.5 g soil of each sample was thoroughly mixed with ml water. The pH was measured with a digital pH meter (Shanghai Lei-ci Co. Ltd) [26]. Temperature and other soil parameters were collected from the public database of China meteorological administration (Table 1).
2.2. Soil DNA Extraction
Soil DNA was extracted from 0.5 g soil of each sample, and the soil was prewashed with 1 ml of 0.5 M EDTA to remove organic matter in the soil [27, 28]. The soil mixture was collected by centrifugation at 12000 rpm for 5 min. Prewashed soil precipitates were further treated with 0.6 ml of 0.5 M CaCl2 and 1.4 ml of ddH2O, and the soil precipitates were collected by centrifugation at 12000 rpm for 5 min [27]. The pretreated soil was lysed with 1 ml DNA extraction buffer (100 mM Tris–HCl, 100 mM EDTA, 100 mM sodium phosphate, 1.5 M NaCl, and 1% (w/v) cetyltrimethylammonium bromide, pH 8.0), 2 μl proteinase K (20 mg/ml) and 200 μl of 20% SDS under the incubation at 65°C for 2 hours. The crude lysate was centrifuged at 17000 g for 10 min and the supernatant was collected. The DNA in the supernatant was purified with the equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) for two times and chloroform:isoamyl alcohol (24:1) for one time. The final supernatant after purification was precipitated with 0.6 volumes of isopropanol, and the soil DNA were collected by centrifugation at 12000 rpm for 5 min. The DNA was dissolved in 30 μl TE buffer with 2 μl RNase (10 mg/ml), and RNA was removed by incubation at 37 °C for 30 min [29].
| Samples | Location | Habitats | Altitude (meters) |
Mean Annual Precipitation (mm / year) |
pH |
Average Temperature (°C) |
|---|---|---|---|---|---|---|
| HN-S1 | Xincai County | Wheat | 40 | 926.7 | 6.97 | 15.0 |
| HN-S2 | Xincai County | Wheat | 40 | 926.7 | 6.76 | 15.0 |
| HN-S8 | Xincai County | Riverside | 40 | 926.7 | 7.66 | 15.0 |
| HN-S9 | Xincai County | Riverside | 40 | 926.7 | 7.35 | 15.0 |
| HN-S10 | Xincai County | Pig Farm | 40 | 926.7 | 6.24 | 15.0 |
| HN-S11 | Xincai County | Pig Farm | 40 | 926.7 | 7.54 | 15.0 |
| HN-S12 | Xincai County | Pig Farm | 40 | 926.7 | 6.69 | 15.0 |
| HN-S13 | Zhengzhou City | Grass | 105 | 632.0 | 7.81 | 14.3 |
| HN-S14 | Zhengzhou City | Grass | 105 | 632.0 | 7.72 | 14.3 |
| HN-S15 | Zhengzhou City | Grass | 105 | 632.0 | 7.79 | 14.3 |
| HN-S18 | Zhengzhou City | Riverside | 105 | 632.0 | 7.59 | 14.3 |
| HN-S19 | Zhengzhou City | Riverside | 105 | 632.0 | 7.75 | 14.3 |
| HN-S21 | Zhengzhou City | Riverside | 105 | 632.0 | 7.74 | 14.3 |
2.3. 16S rDNA gene Fragment Amplification and Soil Microbial Community Analyses
The V3-V4 regions of microbial 16S rDNA genes were amplified with primers of 341F (5’-CCTAYGGGRB GCASCAG-3’) and 806R (5’GGACTACNNGGGTATCT AAT-3’). The 25 µl PCR amplification mixture contained 25 ng DNA, 1 µl each primer (10 µM), 0.5 µl dNTP (2.5 mM), 12.5 µl 2* Vazyme Phata max buffer, 0.5 µl Vazyme Polymerase (Vazyme Biotech). The PCR was performed with an initial denaturation (5 minutes at 95°C), followed by 27 cycles of 15 seconds at 95°C, 15 seconds at 55°C, and 30 seconds at 72°C, and final with one cycle of 5 min at 72°C. The PCR products were purified with the KAPA Pure Beads (Roche) according to the manufacturer’s instructions and further sequenced with an Illumina Miseq system (Illumina).
The raw reads were processed with Usearch fastq_filter, and the low-quality sequences were removed with the default parameters and the data were further analyzed as described before with the Usearch software [30]. The operational taxonomic unit was classified based on 97% identity. The principal coordinates analysis (PCoA) and Non-metric Multidimensional Scaling (NMDS) analyses based Unweighted UniFrac distance were generated by the Vegan 2.4.2 package in R [31]. The raw reads had been submitted to the NCBI Sequences Read Archieve (SRA) database and the accession numbers were SAMN10602944-SAMN10602956.
3. RESULTS
3.1. Overall Soil Microbial Community Composition
A total of 13 soil samples were collected, and they were assigned to be XC group (HN-S1 to HN-S12) and ZZ group (HN-S13 to HN-S21) (Table 1). For the 13 soil samples, a total of 716,285 high-quality 16S rDNA gene fragments were obtained, and they were classified into 4838 operational taxanomy units OTUs based on 97% identity (Table S1). The average sequence and OTU numbers for each sample were 55099 and 1693, respectively, showing there was a large number of common OTUs distributed in these 13 soil samples. The richness and Chao1 indices of these two groups were similar, indicating most microbes in the soil samples were covered by the 16S rRNA sequencing (Table S2) [32, 33]. The Shannon_2 parameters suggested the diversity in these samples was high. Other indices of Simpson, dominance, equitability and rank abundance hinted that the microbial distribution was not definite evenness and some abundant species were available in the soil samples (Table S2). The microbial richness, Chao1, Shannon_2, dominance and equitability parameters of samples in ZZ group were higher than the corresponding indices in XC group. The microbial Simpson parameter of samples in ZZ group was lower than that in XC group (Table 2).
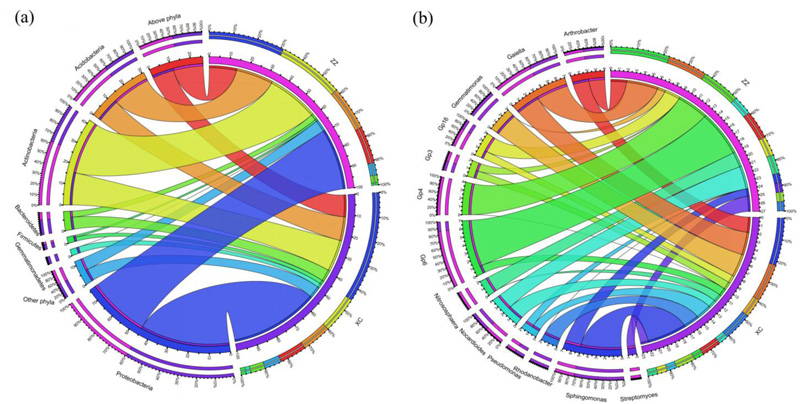
At the phylum level, the most dominant bacteria in the XC soil samples were Proteobacteria, Actinobacteria, Acidobacteria, and Bacteroidetes, and they composed 44.5%, 15.5%, 12.3%, and 6.1%, respectively (Fig. 1A and Table S3); while the most dominant bacteria in the ZZ soil samples were Proteobacteria, Actinobacteria, Acidobacteria, and Bacteroidetes, and they composed 31.1%, 26.4%, 18.5%, and 2.8%, respectively (Fig. 1A and Table S3). According to the previous research, some phyla were classified as dominant phyla in the soil microbiota [34]. At the genus level, 312 genera were identified, and 56.6% of all the sequences cannot be assigned to the known genera, indicating that most bacteria in these soils were unknown (Table S4). Among the assigned genera, the most dominant species were assigned to 13 genera of Gaiella, Sphingomonas, Acidobacteria Gp6, Nocardioides, Arthrobacter, Acidobacteria Gp4, Acidobacteria Gp16, Gemmatimonas, Rhodanobacter, Nitrososphaera, Acidobacteria Gp3, Pseudomonas, and Streptomyces. These dominant genera accounted for 22.7% and 26.5% of the XC group and ZZ group, respectively. Moreover, the distribution of the genera in these two groups was different (Fig. 1B and Table S4). For all the 13 genera, the distribution of the two groups is different, showing soil microbiota of these two places are different (Fig. 1B and Table S4). Especially, the distribution of Sphingomonas, Acidobacteria GP6, Acidobacteria Gp4, Acidobacteria Gp16 and Nitrososphaera between the two groups showed obvious differences.
3.2. Dominant OTUs in the Microbial Communities
Though most microbes in the samples were uncultured, 7 of the 10 most abundant OTUs showed > 97% identities with isolated microbes, suggesting the function of these OTUs can be predicted from the known isolates (Table 3). OTU-4 is the most abundant identified OTU in the samples, and it is Sphingomonas limnosediminicola, which mainly distributed in the wet environment [26]. OTU-1 showed 100% identity with Pseudarthrobacter phenanthrenivorans, which is isolated from a creosote-contaminated soil [27]. OTU-3 was the 3rd most abundant OTU distributed in the soils; it composed 15.94% of microbes in HN-S2 [28]. OTU-17 is Rhodanobacter spathiphylli, which was firstly isolated from a compost-amended potting mix [29]. OTU-9 is Bradyrhizobium namibiense, which is a nitrogen-fixing bacterium [30]. OTU-44 is Nocardioides mesophilus, which is firstly isolated from soil [31]. OTU-94 is Sphingomonas aquatilis, which is widely distributed in the environment.
| Region | Richness | Chao1 | Shannon_2 | Simpson | Dominance | Equitability |
|---|---|---|---|---|---|---|
| XC | 1386.3 ± 300 | 1388.64±300 | 7.92 ± 0.63 | 0.018 ± 0.01 | 0.98 ± 0.01 | 0.76 ± 0.06 |
| ZZ | 2050 ± 265 | 2051.73±264.6 | 9.04 ± 0.34 | 0.0054 ± 0.002 | 0.99 ± 0.002 | 0.82 ± 0.02 |
| OTU | Average composition |
Closest uncultured bacteria (Accession number) |
Identity |
Closest cultured bacteria (Accession number) |
Identity |
|---|---|---|---|---|---|
| OTU_4 | 2.93% | Uncultured Sphingomonas sp. clone DM8-116 (KC172330.1) | 100% | Sphingomonas limnosediminicola 03SUJ6 (NR_157773.1) | 99% |
| OTU_1 | 1.93% | Uncultured Actinobacterium clone 89_2_42 (MH478460.1) | 100% | Pseudarthrobacter phenanthrenivorans Sphe3 (NR_074770.2) | 100% |
| OTU_3 | 1.33% | Uncultured Xanthomonadaceae bacterium clone T92F04c (HM447944.1) | 99% | Chujaibacter soli KIS55-21 (NR_145539.1) | 98% |
| OTU_19 | 1.10% | Uncultured Laceyella sp. clone strain KCTC 3666 (MK121196.1) | 100% | Dehalogenimonas alkenigignens IP3-3 (NR_109657.1) | 86% |
| OTU_17 | 1.08% | Uncultured Gammaproteobacterium clone S1-051 (KF182794.1) | 100% | Rhodanobacter spathiphylli B39 (NR_042434.1) | 99% |
| OTU_9 | 0.96% | Uncultured Alphaproteobacterium(LC378491.1) | 100% | Bradyrhizobium namibiense 5-10 (NR_159233.1) | 100% |
| OTU_44 | 0.94% | Uncultured microorganism clone SGGSWU35(KX925255.1) | 100% | Nocardioides mesophilus MSL 22 (NR_116027.1) | 100% |
| OTU_2 | 0.89% | Uncultured Chitinophagaceae bacterium clone 516_28 (MF002164.1) | 99% | Flavitalea antarctica AQ6-291 (NR_157626.1) | 94% |
| OTU_5 | 0.88% | Uncultured bacterium clone OTU_7933 (MH531581.1) | 100% | Dongia soli D78 (NR_146690.1) | 95% |
| OTU_94 | 0.80% | Uncultured bacterium clone DMA-B01-29(KU886630.1) | 100% | Sphingomonas aquatilis NBRC 16722 (NR_113867.1) | 99% |
3.3. Microbial Diversity in Different Soil Samples
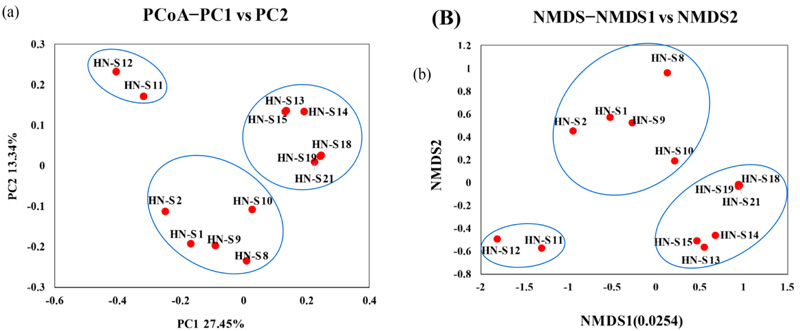
PCoA analyses based on Unweighted UniFrac distance showed that 6 soil samples of HN-S13, HN-S14, HN-S15, HN-S18, HN-S19 and HN-S21 in ZZ group were clustered together (Fig. 2A). Meanwhile, HN-S11 and HN-S12 in XC group were clustered, and another 5 soil samples in the XC group formed the third cluster. The NMDS based on Unweighted UniFrac distance also indicated similar results. The 6 soil samples in ZZ group were clustered together, and another 7 soil samples in the XC group were clustered in two different areas (Fig. 2B). Both PCoA and NMDS presented consistent beta diversity between groups. Besides, the distance between 6 soil samples in ZZ group and 5 soil samples HN-S1, HN-S2, HN-S8, HN-S9 and HN-S10 were close in PCoA and NMDS analyses.
4. DISCUSSION
The dominant bacteria in these 13 soil samples are similar to previous soil microbiota investigations in that the dominant bacteria in soils are Proteobacteria and Actinobacteria [2, 18, 35]. However, the microbial diversity in these 13 soil samples collected from North China Plain is different from the microbial diversity in the East European plain where the most dominant bacteria are Actinobacteria (46.5%) and Proteobacteria (25.6%), it might be due to the fact that the environmental factors between them are distinct [22]. Moreover, the samples collected from the same place, especially samples from XC group, were not completely clustered at the phylum level, hinting even the microbial communities in the same area with different environmental factors were slightly different (Fig. 1A).
More than 50% of sequences cannot be assigned to known genera, suggesting most species in soils were uncultured and investigation of upper layer soil microbes were valuable [2, 18]. The abundance of Sphingomonas genus, which has the ability to metabolize some pollutants, is higher in soils of XC group than in soils of ZZ group, hinting the pollutants in XC are higher than ZZ group. This might be due to the livestock breeding and other agriculture activities in the rural area (XC) group [36, 37]. Bacteria from Gaiella genus can reduce nitrate to nitrite, and its distribution in all these two groups are abundant [38], hinting that these samples might contain a high-level of nitrate. The Rhodanobacter genus can convert nitrate to nitrogen, and its distribution in HN-S1 and HN-S11 are higher than in other samples [39, 40]. This might be due to the fact that a large amount of nitrate was fertilized in HN-S1 and a large amount of nitrate was available in HN-S11, which might derive from pig manure. Besides, the distribution of Nitrosophaera in ZZ group is higher than that in XC group, this might be due to the fact that some nitrogenous fertilizers were added to the soil samples collected in ZZ group.
Most OTUs in the soil samples showed <97% identity with isolated bacteria (Table 3), indicating most species were uncultured. Among the top 10 dominant OTUs, OTU-1 is able to metabolize phenanthrene, suggesting there might be some phenanthrene in the soils of HN-S8 which harbored high-level of OTU-1 [41]. OTU-17 is very abundant in HN-S10, HN-S11 and HN-S12 which sampled from a pig farm and is related with compost, showing this OTU might be functioned in pig manure pollution removal. Some identified OTUs, including OTU-9 and OTU-94, are correlated with soil nutrient cycling and contaminant removal [36, 42], and it might be due to the availability of small amounts of pollutants in the soil samples.
The PCoA and NMDS analyses showed consistent sample classification based on Unweighted UniFrac distance, suggesting the sample classification based on the microbial community was reliable. The bacteria in the ZZ group were more abundant than in the XC group, suggesting that human activities in metropolis increased microbial diversity [24, 43]. The big differences between HN-S11, HN-S12 and another 11 soil samples might attribute to that the input of pig manure from HN-S11 and HN-S12 changed soil nutrition. The microbial distribution of HN-S10 was different from that of HN-S11 and HN-S12, as the pig farm had been abandoned for a few months before we sampled HN-S10, suggesting that the potential pig manure effects on soil microbial distribution had disappeared [15]. As the pH and precipitation of all the samples are nearly the same (Table 1), the soil microbiota of ZZ group and XC group except HN-S11 and HN-S12 are similar, despite the distance between ZZ group and XC group is 300 kilometers. This soil microbiota similarity demonstrates similar pH and precipitation might result in similar core microbiota [11, 13, 19, 44].
CONCLUSION
In summary, we investigated the microbial diversity of 13 soil samples collected from North China plain and found that Proteobacteria, Actinobacteria, Acidobacteria, and Bacteroidetes were the dominant bacteria. Moreover, the microbial species in the North China plain was similar, but the microbial distribution was different, indicating different area would have a different core microbiome. Input of nutrition, such as pig manure of HN-11 and HN-12, to the soil would change soil microbial distribution, showing environmental factors are the key ecosystem driving roles for microbial distribution.
AUTHOR'S CONTRIBUTIONS
Conceptualization, Y.W.; methodology, Y.W., L.W., Q.Z; software, B.J.; validation, L.W., Q.Z., H.M., X.C., M.W., Y.Z. and Y.W.; formal analysis, B.J., Y.W.; investigation, L.W., Q.Z., H.M., X.C., M.W., Y.Z.; resources, X.X.; data curation, X.X.; writing—original draft preparation, L.W., Q.Z., X.C., M.W., and Y.Z.; writing—review and editing, B.J., Y.W; visualization, Y.W.; supervision, Y.W.; project administration, Y.W.; funding acquisition, Y.W., L.W., Q.Z., H.M., X.C., M.W., and Y.Z.. All authors have read and agreed to the published version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
DATA AVAILABILITY STATEMENT
The raw reads of the 16S rRNA data had been submitted to the NCBI Sequences Read Archieve (SRA) database and the accession numbers were SAMN10602944-SAMN10602956.
FUNDING
This research was funded by Zhengzhou University startup foundation (No. 32210876) and Innovation and Entrepreneurship Training Program for College Students of Zhengzhou University (2018cxcy717), and Henan Shangxing Environmental protection Technology Co. LTD.
CONFLICT OF INTEREST
Dr. Yongjun Wei is the Editorial Advisory Board Member of the journal The Open Microbiology Journal.
ACKNOWLEDGEMENTS
We thank DeepBiome Co., Ltd. for bioinformatic assistance.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

