All published articles of this journal are available on ScienceDirect.

Applications of Metagenomics for Unrevealing the Extended Horizons of Microbiota Prevalence from Soil to Human Health
Authors Info & Affiliations
Abstract
Phylogenetic analysis of different ecosystems has shown that the number of microbial communities in a single sample exceeds their cultured counterparts. Microbes have been found throughout nature and can thrive in adverse conditions. Besides inhabiting diverse environments, they also play a key role in the maintenance of the ecosystem. Most of these microbes are either unculturable or difficult to culture with conventional culturing methods. Metagenomics is an emerging field of science that has been in the light for a decade and offers a potential way to assess microbial diversity. The development of metagenomics opens new ways to study genetic material directly from the environmental samples. DNA sequencing and synthesis technologies are making it possible to read and write entire genomes. The huge amount of data obtained from genome sequencing inevitably requires bioinformatics tools to handle and further process them for analysis. Advances in DNA sequencing and high-performance computing have brought about exemplar improvement in metagenomics, allowing in-depth study of the largely unexplored frontier of microbial life. This culture-independent method provides extensive information regarding the structure, composition, and function of the diverse assemblages of the environmental microbes. The current review presents an overview of the technical aspects of metagenomics along with its diverse applications.
1. INTRODUCTION
Microbes are the most diverse, ubiquitous, and profuse forms of life that make up the major portion of biomass (≈70 Gt C) (1 Gt C = 1015 g of carbon) on Earth, of which bacteria constitute 70 Gt and Archaea comprises of 7 Gt [1]. This unseen majority plays an important role in ecosystem functioning [2] and human health [3]. It co-exists in communities, where its determined actions have significant contributions in sustaining the life on the planet. Considering the unprecedented role of microbes in the health and disease status in humans, the microbiome is considered the last organ of the body [4]. Numerous studies indicated a high degree of correlation between the human gut microbiome and the health and disease status of an individual, with the human gut microbiome influencing human wellbeing through immune function, physiology, nutrition, and metabolism [5].
Consequently, characterization and understanding of the human gut microbiome, along with its correlation with diseases and well being of the individual’s health, has fascinated global researchers and forced them to explore the unseen majority inhibiting the gut. Studies confirmed that the human gut microbiome consists approximately 15,000 to 36,000 bacterial species [6]. The gut microbiome is estimated to contain 100 times more genes, as compared to 25,000 genes in humans [7]. Accordingly, the identification and characterization of the gut microbiome is the major focus of microbiome research. However, it has been estimated that approximately 95% of the microbial communities are non-culturable by the currently available techniques [8]. These uncultured microbes are untapped reservoirs of novel biomolecules such as enzymes, drugs, as well as metabolic capabilities. Moreover, large microbial populations are also present in/on animals, including humans.
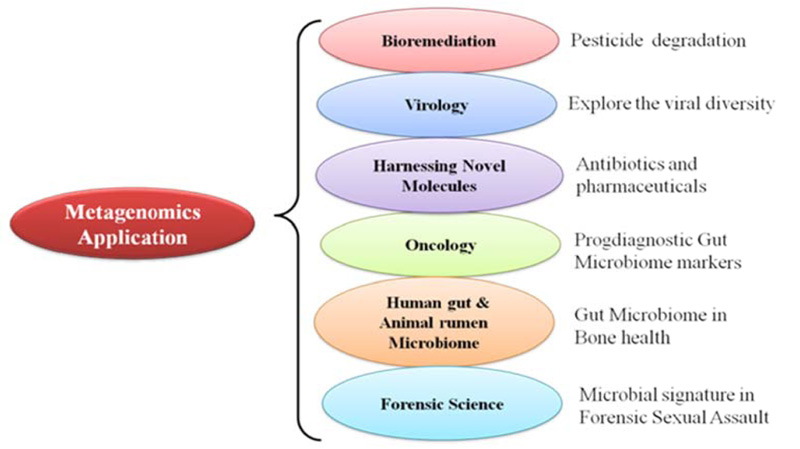
However, these microbiotas have been found to play a crucial role in maintaining normal physiological well-being. Considering the possible role of microbiome in diverse fields, DNA sequencing is a promising technology to understand and explore the potential of this unseen diversity. Furthermore, microbiome studies have changed the way in which microbiologists have approached various problems. It has redefined the perception of the genome and also hastened the rate of gene discovery. The multiple spectra of microbiome applications have been represented in Fig. (1).
2. EXPLORING MICROBIOME FUNCTIONS
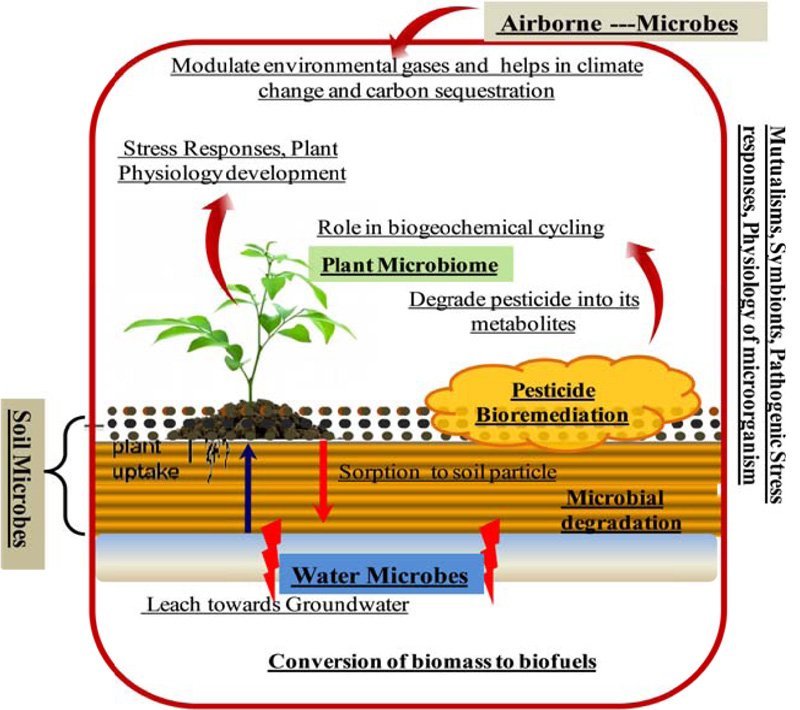
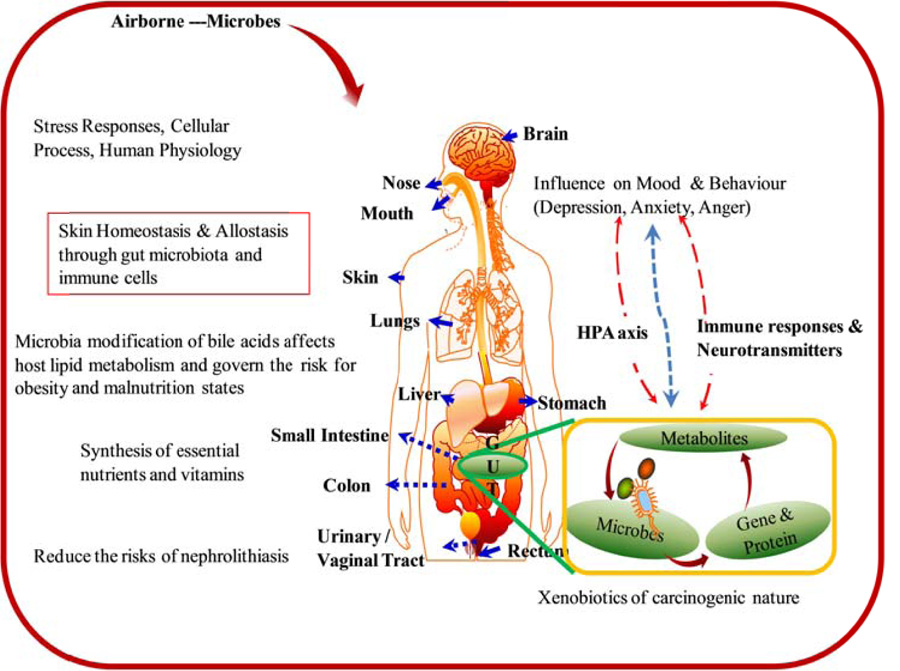
Exploring the structure, dynamics, and functional attributes of different microbial communities associated with different niches has become a major challenge for microbiologists. However, the recent advances in microbiome research have significantly helped global scientists to overcome these challenges. Understanding microbiome functions can enable the engineering of some novel diagnostic approaches and interventional strategies that can be used in a diverse spectrum of fields, spanning from medicine and human health to environment, agriculture, and ecology to forensic science [9]. Moreover, microbiome research is expected to address some of the most vexing, inspiring, and fundamental questions of 21st-century research. Overview of the prevalence of microbiome in the environment (Fig. 2) and their interaction with humans is given in Fig. (3).
Some of the key examples of functional contributions of the microbiome are as under:
(1) Xenobiotics Metabolism: The microbiome functions as an unexplored regulator of xenobiotic metabolism. Microbial communities serve as the potential alternatives for the elimination of toxic pollutants from the environment [10]. The gut microbiome is found to carry out the detoxification of ingested xenobiotics of carcinogenic nature [11]. Similarly, the oxalate metabolism by microbial taxa (Oxalobacter formigenes) inhabiting the gut reduces the risks of nephrolithiasis [11]. Studies suggested that the microbial modification of bile acids affects host lipid metabolism [4].
(2) Harvesting of Inaccessible Nutrients: Microbial communities occupying the gut have been found to aid in the synthesis of essential nutrients and vitamins and in harvesting the otherwise inaccessible nutrients from the diet [12]. The digestive capacity of an individual depends on its microbiota [12]. The gut microbiome also plays an important role in identifying the host individuals at risk for obesity and malnutrition states and treating these nutritional disorders based on host-microbial ecology.
(3) Gastrointestinal Cell Renewal: The gut is one of the highly regenerative organs in the human body. Persistent renewal of epithelial cells allows the gut epithelium to withstand the wear and tear that it undergoes during digestion and eliminating waste. However, in the case of germ-free animals, the renewal is slower than the conventional ones, the reason being the absence of bacterial flora [13]. Both the skin and gut are the most richly innervated and densely vascularised organs, both exclusively related in purpose and function, and possess the most vital neuroendocrine and immune roles [14]. Considering our primary interface with the external environment, both skin and gut are essential body organs to the maintenance of physiologic homeostasis.
Evidence suggests a bidirectional and intimate connection between the skin and the gut, with several studies linking gut health to skin homeostasis and allostasis (Fig. 3) [14]. All these processes influence in parts via interactions between microbiota and immune cells [15] and have a series of effects ranging from susceptibility to neoplasia [15] to the renewal of the damaged mucosal barriers [16].
3. ROLE OF GUT MICROBIOME IN HUMAN HEALTH
3.1. Neuropsychological Disorder
Depression is one of the major debilitating psychiatric illnesses [17] and is estimated to affect more than 300 million people worldwide [18]. Major depressive disorders are one of the leading causes of global disability and account for more than ~800,000 suicide deaths annually [18]. Irrespective of some of the significant advances in understanding the etiology of major depressive disorders, it is believed that the present status of the knowledge is incomplete, treatments are scarce, and novel insights into these disorders are urgent and need of the hour. Unfortunately, pathophysiologies of the major depressive disorders in association with the gut microbiome are not clear (Fig. 3). Just a decade ago, the idea that the gut microbiome could have a bearing on the human brain was often considered wild and unlikely to happen.
The advancements in sequencing technologies and informatics pipeline analysis unlock the connection that the gut microbiome influences mood and prevents depression (Fig. 3). Links between CNS and the trillions of the microbes colonizing the human gut and gut microbiome are now a major focus of present-day medical research [19]. However, the mechanism by which the microbes govern brain functioning linked to altered behaviour and memory is still in its infancy and needs to be explored. Genomic surveys of the gut inhabitants have revealed that these diverse microbial communities play a part in programming the hypothalamic-pituitary-adrenal axis along with the stress reactivity over the life span (Fig. 3) [20]. Functionally, the stress responses are immature at birth and develop throughout the postnatal period, corresponding to gut bacterial colonization.
Earlier several animal studies evidentially reported that stress can perturb the composition of the gut microbiome and that enteric pathogens might affect host behaviour by mediating the immune responses and neurotransmitters [21]. Studies suggested that significant behavioral differences in the brains of animals with altered or absent gut microbiota displayed various molecular differences [9]. These included brain-region-specific changes in levels of Brain-Derived Neurotrophic Factor (BDNF), differences in the expression of various neurotransmitter receptors like catecholamine’s, serotonin and alterations in the turnover of certain neurotransmitters such as glutamate and γ-Aminobutyric Acid (GABA) [21]. Recently, researchers demonstrated that certain species of bacteria had been found to be associated with depression and quality of life. Moreover, the metabolic products of gut microbiota are also linked to measures of mental health, psychiatric illnesses, and the personality of humans [22].
3.2. Role of Gut Microbiome in Bone Health
The human body is home to trillions of microbes that differ in composition across different anatomical sites of the body [23], with Bacteroidetes, Firmicutes Actinobacteria, Fusobacteria, and Proteobacteria being the most dominant phyla [24]. However, the relative proportions of these taxa vary between individuals and different sites within the body. Studies reflect that the microbial communities inhabiting one part of the body significantly differ from the other part [25]. It is believed that only a small fraction (up to 10%) of the microbes are similar among different individuals, with adults possessing a relatively stable microbiome. It is a fact that the gut microbiota plays an important role in digestion, metabolism regulation, immune protection and can affect growth, reproduction, and the overall dynamics of an individual (Fig. 3) [26]. The link between Gut Microbiome and bone health is recognized for the first time by Viney and Riley, 2014 [27]. Further researchers have also suggested the manipulation of the gut microbiota and its association with bone health [28].
The relationship between the gut microbiota and bone health is complex and involves various processes such as the regulation of immunomodulation (CD4+ T cell activation), regulation of key bone minerals (calcium and phosphorous), and translocation of essential microbial products across gut endothelium (peptidoglycans, lipopolysaccharides, and short-chain fatty acids) [21, 22]. For the first time, investigations carried out by Sjögren et al. [28] showed that bone mass and density are regulated by the gut microbiome. Sjögren et al. found that the female germ-free mice of 9 weeks of age had a higher cortical and trabecular bone density, compared to the control mice who rose in the conventional conditions. Likewise, studies conducted on mice models have concluded that different strains of bacteria (Bifidobacterium and Lactobacillus) have the potential to increase the bone mineral density trabecular number [28, 29].
On the other hand, regulatory T cells (Tregs) are responsible for the expression of the transcription factor FoxP3 and are capable of blocking the proliferation of the conventional T cells and effector cytokines [30]. It is evident from too many studies that bone health is largely affected by the deficiency or dysbiosis of the GM [28, 29]. So, some logical strategies must be there to overcome this menace and to minimize the GM alterations with the beneficial ones. Probiotics, a class of symbiotic microbes, is believed to have a potential effect on bone build-up. The possible mechanism of this process may occur via microbial synthesis of different enzymes, metabolites, and vitamins C, D, or K or folate [31].
In contemporary prebiotics, the oligosaccharides, i.e., polydextrose, fructose and galactooligosaccharides, xylo-oligosaccharides, and soybean oligosaccharides are also used for bone health, but their beneficial effect is still unclear. These oligosaccharides reveal their beneficial effect only in their calcium absorption in animals and postmenopausal women [32]. As in this case, the dietary fiber is fermented to Short-Chain Fatty Acids (SCFAs) in the lower region of the gut, thereby increasing the pH thus ultimately increasing the calcium absorption [33]. These SCFAs also aid in the energy requirement of the gut epithelial cells, which in turn may improve gut permeability and health [34].
All these studies recommend a detailed understanding of the Gut Microbiome dysbiosis and its effect on the skeletal system [28, 29, 31]. These interventions also suggest some dietary modifications affecting the Gut Microbiome that will establish potential therapeutic strategies for promoting bone health. These findings suggest that the oral administration of probiotics can significantly reduce bone loss.
3.3. Cancer and Gut Microbiome
At present, microbiome studies are receiving significant attention given their influence on human health and diseases, including cancer. The role of the microbiome is becoming apparent with evidence mounting in supporting its role in cancer treatment and is recognized as an important factor associated with tumour development and the effectiveness of anticancer therapies. Gut microbiome has evolved as a key player in maintaining human health, influencing not only the gastrointestinal tract but also the distal organs such as the liver, pancreas, brain, etc [35, 36]. Therefore, the compositional and the functional alterations of the gut microbiota (dysbiosis) contribute towards the development of various pathological conditions, including various cancers. Bacteria have also been reported to elevate inflammation and cancer cell proliferation via the production of cytokines.
The microbiomes have been found to have a close association with cancer via direct or indirect pathways. The direct routes include the interaction of pathogenic microbial communities with the immune system via bacterial antigenic particles with pattern recognition receptors (e.g., toll-like receptor) or through direct colonization of epithelia by pathogenic bacteria. In contrast, the indirect pathways include the microbial production of carcinogens and chemoprotective factors from endogenous sources, such as compounds resulting from human metabolism, or from exogenous sources, such as diet [37].
Recently, the faecal microbiome analysis from patients with colorectal cancer identified colorectal cancer-enriched bacteria. These bacteria include Alistipes, Bacteroides, Fusobacterium, Parvimonas, Porphyromonas Prevotella, and Thermanaerovibrio and serve as potential diagnostic bacterial markers across the populations [38, 39]. Correlation of colorectal cancer-enriched bacteria with various biosynthesis, metabolic, and degradation pathways provides key insights into the functional capacity of these bacteria in colorectal cancer [39]. These intestinal bugs also influence the incidence and progression of extra-intestinal cancers, including hepatocellular carcinoma and breast cancers, probably via inflammatory and metabolic circuitries [40, 41]. All these studies are compatible with the findings from epidemiological studies that show a link between microbial dysbiosis, along with its consequences [42, 43]. In addition, the faecal microbiome studies not only establish a link between the gut microbiome and cancer but also help in identifying the key microbial signatures that can precisely predict the disease across the populations.
The gut microbiota is believed to affect oncogenesis and tumour progression both locally as well as systemically, with the support of inflammatory and metabolic cues. Specific bacterial taxa inhabiting the gut are found to affect the cancer treatments by modulating the host immune response and through drug metabolism.
Multiple studies have shown gut microbiome signifying its influence on the response to immune checkpoint blockade across different cancer types [44]. The crosstalk between the immune system and gut microbiome is widespread and critical. It does not only permit the tolerance to oral food antigens or the commensal microbes but also facilitates the immune system to recognize and attack the opportunistic bacteria, thus averting both bacterial invasions as well as an infection.
Disturbance in the balance of commensal microbes results in dysbiosis, which is characterized by a less stable and diverse beneficial microbiota and with potential enrichment pathogenic bacteria [45]. Dysbiosis is related to a plethora of diseases, including skin to neurological disorders and from inflammatory to metabolic dysfunction. Moreover, recent findings have shed light on a number of diseases directly impacted by the gut microbiome. Therefore, the gut microbiome is becoming a key factor in understanding and treating complex diseases such as Crohn’s disease, obesity, type 1 and type 2 diabetes, rheumatoid arthritis, and atherosclerosis [4].
4. MICROBIOMES FOR HARNESSING NOVEL MOLECULES
Soil constitutes one of the complex environments of the terrestrial ecosystems and is one of the major and key pools of microbial diversity [46]. The diversity of microbes in these soil ecosystems exceeds every other environment like the gut, rumen, forage plants, etc., with an estimated number of about 1030 [47]. Among these, prokaryotes (106 to 108) represent the largest proportion [48]. Soil microbial diversity plays an imperative role in soil ecosystems [49]; therefore, a healthy soil microbiome is important for ecosystem functioning. The combination of microbiomes from various ecosystems and niches (soil, gut, water, rhizosphere, etc.) upholds the fabric of life.
Unravelling the tree of life, dominated by bacterial diversifications, is essential to expand our view about the diverse microbiome. Soil metagenomics provides a culture-free assessment of the mostly unexploited genetic pool of soil microbial communities and provides the unique opportunity to explore soil’s limitless environments harbouring scores of yet unknown and mostly unculturable microbes and other organisms. Soil metagenomics allows scientists to peep into the more complex picture of soil microorganisms and to develop an understanding of their interactions. The development of metagenomics and its applications has opened a way to unculturable microbial communities of the soil, thereby availing a rich and prosperous source of some of the novel and valuable biomolecules (Fig. 4).
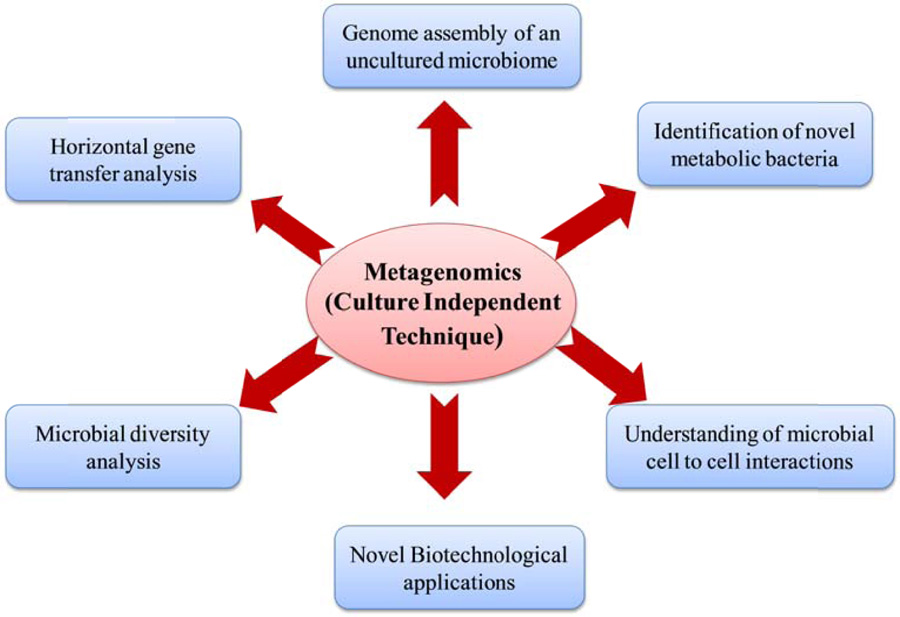
Some examples of the applications of soil metagenomics are as follows:
4.1. Antibiotics and Pharmaceuticals
Soil metagenomics possesses the potential that has significantly affected the production of antibiotics. Studies carried out [50, 51] reported the successful screening of the soil metagenome from Indirubin [52, 53] while analyzing the soil metagenome detected a wide range of novel antibiotics. The same authors, while screening seven different metagenomic libraries from soil, stated that these libraries uncovered 11 clones producing long-chain N-acyltyrosine antibiotics, and their synthetic analysis showed that ten of these antibiotics were novel.
4.2. Oxidoreductases/Dehydrogenases
Oxidoreductases are the enzymes that are capable of oxidizing the short-chain polyols, thereby serving as useful biocatalysts in the industrial production of amino acids, alcohols, hydroxyl acids. Representing diverse range of enzymes, Oxidoreductases can broadly be divided into four major groups; (i) oxidases, (ii) oxygenases/hydroxylases, (iii) dehydrogenases/reductases, and (iv) peroxidases [54]. These enzymes are responsible for catalyzing a diverse range of reactions such as hydroxylation, dehalogenation, deamination, oxidation, dealkylation, dehydratation, dehydrogenation, epoxidation, and reduction.
Recent investigations into Oxidoreductases isolation have largely focussed on the terrestrial settings; however, the extremes of oxygen, temperature, pressure, and substrate availability presented by aquatic environments have also encouraged their exploration as a potentially rich source of novel biocatalysts. Sequence based metagenomic approaches have played a key role in identifying and exploring the potential marine ecosystems candidate sources for novel enzymes [55].
4.3. Vitamins Biosynthesis
Soil metagenomics has found its role in searching for the novel genes that code for the synthesis of several vitamins like biotin [56, 57]. Advances in molecular biology methods, particularly the next-generation high-throughput sequencing, omics and Insilco analysis approaches, have allowed us to access and explore the vast microbial diversity of biotechnological importance and has made it possible to identify and explore novel biological molecules with specific activities without the need for isolation and culturing of microbes [58].
4.4. Polysaccharide Degrading Enzymes
Cellulases possess several applications and have substantial biotechnological potential in many industries such as fuel, food, and chemicals. Amalyses have been a key and important focus of several metagenomic studies, with a number of reports available regarding the purification and characterization of the novel amylolytic enzymes [59].
4.5. Amidases
Amidase-positive clones were first detected through [60, 61] analyzing the soil metagenomic library for various biocatalysts. Amidases are used in the biosynthesis of 9-lactam antibiotics. Pan et al. [62], while targeting the amidases, detected seven positive amidase clones, one of which was a novel penicillin acylase.
5. METAGENOMICS: A WAY FORWARD TO EXPLORE THE VIRAL DIVERSITY
Recent development and cost reduction in sequencing approaches have revolutionized virology with the discovery of many novel viruses and viroids [63]. Viruses can be identified via a broad array of techniques that mostly depend on the contrast with the ones that are already known. Some of these historic methods include cell culture, electron microscopy, and inoculation; however, these methods show limitations. For example, a number of viruses cannot be cultured, thereby excluding the use of cell line isolation and serologic methods, and these can only be characterized using different molecular methods. Ouborg et al. [64] summarized different molecular techniques viz. PCR-based and subtractive hybridization-based methods, and microarray to classify new viruses. Although these techniques have been endorsed by the scientific world with the discovery of many viruses, they still require the knowledge of similar viruses.
The recent advancement in sequence-independent PCR-based methods has helped to overcome these curbs. Many viruses, in particular RNA or single-stranded DNA viruses, exhibit extreme evolutionary dynamics. They have varied high mutation rates, up to six orders of magnitude higher than in humans, short generation times, and large population sizes. Under these conditions, genetic variants are produced constantly, and each infected host-virus population displays a higher degree of genetic diversity. With the introduction of NGS technologies, the experimental analysis of genetic diversity has changed dramatically.
From the past decade, a number of studies while using metagenomics techniques have offered a substantial amount of information regarding the identification of novel viruses from human samples. A number of infectious viral diseases were recognized before the identification and classification of their causal agents. With the steady increase in the growth of the viral metagenomics, a number of noble viruses have been isolated in a relatively short period associated with the disease outcome in humans [64, 65]. Viruses viz. Arena virus, Paralysis virus, Lujo virus, Borna virus, Simian hemorrhagic fever virus, and Klasse virus are the notable novel viruses that have been identified by metagenomics approaches as causative agents of diseases in humans and other mammals [66].
Recent studies have offered significant information related to the identification of a number of viruses associated with public health [67]. Studies like these have high lightened the future perspectives of the metagenomics approaches to generate a huge amount of data for the identification of the unidentified and potent infectious agents of humans within a relatively short period. Overall, metagenomics has provided considerable insight into host-virus interaction and the overall viral diversity in different and varied environments.
6. ROLE OF METAGENOMICS IN EXPLORING THE RUMEN MICROBIOME
Livestock production is a subsidiary of agricultural production throughout the world, with India being no exception. Ruminants in tropical countries are mainly fed on lignocellulosic by-products of agricultural origin like straw, tree foliage, and oilseed cakes. Digestion of these plant materials in ruminants occurs with the help of extensive microbial communities, including bacteria, fungi, and protozoa [68], which are present in the rumen and endow the host with nutrients. Studies carried by Lauck et al. [69] proved that the microbes present in the rumen belong to three domains viz., Bacteria, Archea (methanogens), and Eukarya (fungi and protozoa). The rumen microbiome is a source of the unique microbial diversity that is the source of plant cell wall-degrading enzymes.
The advent of the next-generation sequencing technologies has made it possible for researchers to describe rumen microbial diversity at a much greater resolution than that of the previous studies [70]. Rumen fluid serves as an excellent source for the mining of CAZymes mainly because of its apparent choice for and evolution as a complex lignocellulosic degradation system [71]. However, identification of these enzymes is restrained because most of the microbes that inhabit the rumen are unculturable. The identification of candidate carbohydrate-active genes from metagenomic sequence data sets initially was a de novo assembly performed with the raw sequences followed by a prediction of the putative open reading frame. This study offers new insights into the field of microbiology as well as for biotechnology. Data generated from the rumen metagenome may be worked upon for isolating the potential enzymes of industrial importance.
7. BIOREMEDIATION: A METAGENOMICS DRIFT
Since the day humans invaded the planet, it has been packed with several toxic pollutants from different sources. Recent advancements in scientific technology have reduced environmental pollution in a number of ways, and bioremediation is considered as one of the finest ways to neutralize contaminated environments [72]. The emergence of the “omic” approaches has the potential and promises to revolutionize the field of microbiology by offering direct access to microbial communities inhabiting the contaminated environments [73]. The metagenomics approach has best been recognized as an efficient method for the removal of various types of pollutants [74].
Several studies carried out [75] suggested that microbial communities serve as the potential alternatives for the elimination of toxic pollutants from the environment (Fig. 2). These molecular techniques have been applied in monitoring the bioremediation process of the hydrocarbon, and associated pollutants and invariably have significantly improved the biological treatment of these contaminants [76]. The increased applications of the metagenomics techniques have narrowed down the gap and are reported to be used in exploring the diversity of the many activated sludge habitats [77]. Precise indication regarding the analysis of microbial diversity of hydrocarbon polluted habitats has been stated in oil leak-affected mangrove areas of Brazil, the sediment of the deep-sea of Mexico, and in petroleum sewage [78]. Besides its role in analyzing the taxonomic diversity of a particular area, metagenomics also allows us to design a pipeline to chalk out the degradation pathways of a particular area.
Bioremediation for eternity has accommodated newer advances of science and technology for the establishment of better environments. Some studies carried out [65] confirmed that there had been a gradual rise in the interest in metagenomics-based bioremediation studies. These studies suggest that metagenomics may prove to be one of the best adaptations for bioremediation, therefore leading towards the formation of healthy and non-toxic environments.
8. ROLE OF METAGENOMICS IN THE SYNTHESIS OF BIOFUELS
Renewable energy resources such as biodiesel and bioethanol have gained much of the attention of scientists and industries mainly because of their environmental friendliness, cost-effectiveness, and social benefits. Alternative energy sources based on the conversion of biomass to biofuels require cost-effective techniques so that they are economically realistic. The need of the hour is to screen out some of the novel enzymes (such as 33 Carbohydrate-Binding Modules (CBM33), Glycoside Hydrolases (GH61)), catalyzing the oxidative conversion of biomass to biofuels [79]. According to Leresche et al. [80], bacteria seem to be the main prime source for these (Endoglucanase, Cellodextrinase β-glucosidase, Xylanase Mannanase, Xylosidase, Arbinofuranosidase, Lichenase, Glucanase, Amylase, Phosphophenol pyruvate, carboxykinase, Xylanase, Cyclophilin, Mannanase) enzymes.
Since more than 99% of the microbes are unculturable, a large pool of the genes and gene products (enzymes) remains unexploited. With the rapid development of next-generation sequencing techniques, metagenomics has been employed to identify the enzymes used for biofuels production. This technology facilitated scientists to found out many novel enzymes like endoglucanase, amylolytic enzymes, xylanase, lignases, β-glucosidase for bio-ethanol, and lipolytic enzymes for biodiesel production [81]. These enzymes may have the potential for new applications in biofuels production. To date, the comparative biomass-degrading enzymes that were screened from the environmental samples include polysaccharide-modifying enzymes (cellulases, α-amylases, xylanases, 1, 4-α-glucan branching enzymes), dehydrogenases, oxygenases, Oxidoreductases, carboxyl-hydrolases (esterases, lipases).
9. MICROBIAL SIGNATURE IN FORENSIC SEXUAL ASSAULT CASES
In medicolegal cases like sexual assault (rape), association of the accused with the victim is a challenging task for the medical practitioners as well as forensic experts. Studies have suggested that the microbial communities inhabiting individual’s pubic hairs could function as potential signatures to trace their involvement in sexual assault cases [82]. Hair function as one of the most common types of trace evidence in any forensic investigation; however, the bulk of the hairs received from the crime scene lack their roots and contain less amount of human genetic material to carry out DNA profiling of suspects. In that case, hair overloaded with microbial colonies favours as microbial signatures that could function as an alternative to track out the suspects.
Seminal and vaginal fluid, pubic hairs microbiota profiling after alleged sexual assault favours a clue of association with the victim and accused [83]. The occurrence of seminal, vaginal fluid, saliva, and pubic hairs is often found at the scene of the crime. The microbiota inhibited in biological fluids appeared to be transferred during intercourse, suggesting its potential for forensic analysis on sexual assault cases.
10. MINING THE BIG MICROBIOME DATA
Considering the big microbiome data sets, the use of next-generation high throughput sequencing techniques would be a promising technology for mining it, rather than the conventional cultivation approaches. DNA sequencing, followed by assembly and taxonomic and functional annotation, are the prerequisites for understanding and exploring different microbiomes. However, revealing and unlocking microbiome data sets were quite difficult owing to the limitations inabilities to obtain and process the data generated by sequencing.
Nowadays, next-generation high-throughput sequencing technologies, such as multiple displacement amplification for single-cell sequencing, and numerous statistical analysis tools, such as QIIME [84], MG-RAST [85], Mothur [86], MEGAN [87], PICRUSt [88] for microbiome data, made it possible to unlock the microbial communities from various perspectives. The modern high throughput sequencing approaches in combination with the Insilco analysis tools allow a much detailed and comprehensive study of diverse microbial communities. Integration of all these methods can answer the most fundamental questions of microbiology, i.e., “who is there?” and “what are they doing?” and “how are they doing it?”
11. LIMITATIONS OF METAGENOMICS
Metagenomics, the so-called fourth domain of life, deals with the molecular approach of culturing microbes. However, Metagenomics has established itself as an indispensable and widely affordable tool for studying unculturable microorganisms (Bacteria, Archaea, and viruses) [89]. Metagenomics also has certain limitations. The main drawback of this technique is the lack of knowledge of the environment and its instability with respect to time and space and hence cannot guarantee to sample representatively and reproducibility.
To accumulate knowledge based on ecological niches, there is a need for the development of tools with better descriptive modes. Despite the formidable advancement in high throughput and the cost efficiency of sequencing technologies, the cost of sequencing remained a limiting factor. The functional screening may be unfair because of the use of heterologous systems for the expression of the gene, the reason being that it might have inappropriate gene expression, risk of toxicity, etc. The demand is for the development of new heterologous expression vector systems that may represent the biology as well as the ecology of the phylogenetic division. This calls for a sober investment to work out the cultivability of some of the recalcitrant microorganisms.
CONCLUSION
Uncultured microorganisms comprise the majority of the planet’s biological diversity. Based on their habitat and unique characteristic features, these microorganisms serve as a powerful source of many important metabolites such as antibiotics, enzymes, proteins, etc. Understanding the real value of genetic diversity of the microbial and the increasing pursuits in biotechnological applications of microbial by-products such as metabolites, antibiotics, enzymes, proteins, biomaterials, etc., may incite the development of innovative research techniques for the direct or indirect acquirement of these genes. As outlined, metagenomic analyses facilitate a comprehensive study of the microbial communities along with the unparalleled access of genetic diversity therein. Outrageous information has been achieved in a relatively short period concerning the taxonomy, phylogeny, and genetic novelty among the diverse metagenome. The advent of the new molecular techniques and the continuous decrease in the sequencing rates will bloom this at an increasing rate in the near future.
LIST OF ABBREVIATIONS
| Gt | = Gigatons |
| C | = Carbon |
| CNS | = Central Nervous System |
| GM | = Gut Microbiome |
| SCFAs | = Short Chain Fatty Acids |
| PCR | = Polymerase Chain Reaction |
| BDNF | = Brain-Derived Neurotrophic Factor |
| GABA | = γ-Aminobutyric Acid |
| QIIME | = Quantative Insights into Microbial Ecology |
| MG-RAST | = Metagenomics Rapid Annotation Using Subsystem Technology |
| MEGAN | = Metagenome Analyzer |
| PICRUSt | = Phylogenetic Investigation of Communities by Reconstruction of Unobserved States |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICTS OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors are thankful to the Dr. Harisingh Gour University, Sagar (MP), and CTM-IRTE, Faridabad, India, for providing the opportunity to work and their support and interest.

