All published articles of this journal are available on ScienceDirect.

Improved Production of HIV-1 Subtype C Protease from Transgenic E. Coli
Abstract
Background:
Human Immunodeficiency Virus 1 (HIV-1) subtype C is responsible for the majority of infections of patients in Southern Africa. The HIV protease is a primary target for the development of highly efficient anti-retroviral pharmaceuticals because of its pivotal role in the maturation of the virus in the host cell. For target validation of novel HIV protease inhibitors, there is a need for the availability of an abundance of this protease.
Objective:
This study reports an optimized method to produce HIV-1 protease derived from HIV-1 subtype C.
Methods:
It involves the use of a transgenic E. coli strain that overexpresses the native form of the enzyme via inclusion bodies. A stringent method for the isolation, purification, and renaturation resulted in the production of highly pure active HIV-1 protease. In order to facilitate an increase in protease yields, an optimized growth strategy was developed. In this regard, a chemically defined medium with lower glucose content and devoid of essential amino acids of the TCA cycle was used as an alternative to the widely used nutrient-rich Luria Bertani (LB) medium.
Results:
Results indicated an increase in protease yield up to twice the amount, thereby making this medium an attractive alternative for increasing biomass and HIV protease production for future research.
Conclusion:
An optimized method for HIV-1 protease derived from HIV-1 subtype C production using chemically defined media was established. This was achieved using a known method to isolate and purify the enzyme with the use of a specialized feeding strategy.
1. INTRODUCTION
The genetic variability of HIV is extensive, and because of this, it is classified into many types, sub-types and recombinant forms [1, 2]. HIV is divided into two types, the more virulent and easily transmitted, HIV-1, and the less widespread HIV-2, which has a slower transmission and infection rate [3, 4]. HIV-1 is further categorized into nine sub-types: A-D, F-H, J and K. HIV-1 subtype B being well understood and the most prevalent in North America and Western Europe [5]. HIV-1 subtype C (CSA) is responsible for the majority of infections in the sub-Saharan region of Africa, with a few cases of subtype A and G being reported [5]. In South Africa HIV-1 subtype C accounts for approximately 95% of infections [6]. It has widely been reported that the structure of these subtypes varies quite considerably and as a result, anti-HIV drugs are ineffective due to their genetic diversity [7].
Research regarding HIV-1 protease is mainly with the enzymes that are isolated from HIV-1 subtype B, but little information is known about HIV protease that is isolated from subtype C. It has been reported that although structurally similar, these enzymes isolated from different subtypes differ in their catalytic ability and binding capacity to the active site [5, 8, 9]. Furthermore, mutations of the subtypes create independent drug resistance [10]. Therefore, there is a need for crucial information of the protease that is derived from subtype C to be examined if any progress of its activation is to be made. For this, there is a need for an abundance of this enzyme to be available for inhibitor testing and enzymology. Previously HIV protease has been synthesized chemically [11], but a more common method for isolation includes heterologous protease production in a vector system. Generally, genetically engineered bacteria are used as vehicles to produce these enzymes as inclusion bodies so that it can easily be isolated.
Due to the complex nature of HIV and its dangerous infectious capacity, it is one of the most difficult organisms to work with. Thus, alternative methods for the study of this specific enzyme need to be considered. Escherichia coli (E. coli) has been used extensively in the past and currently as vehicles to express heterologous proteins from wild-type organisms that are not easily accessible [12]. Due to the lack of natural selection, the relationship between E. coli and the protein produced is directly proportional [13] therefore, there is a vast area of interest in creating an environment that enables increased cell density cultures, thereby allowing maximum production of recombinant proteins [14-17]. Studies have shown that manipulation of the nutrient content and feeding strategy of E. coli can result in an increase in target proteins.
A study by Ido et al. [18] created an accessible platform to study HIV-PR using an E. coli vector system for easy isolation of the protease. PCR-based methods incorporated with multiple restriction enzyme reactions were used to create a plasmid that expresses the HIV-PR enzyme when transformed into the E. coli bacterium [18]. This strategy to clone the HIV-PR gene of the virus was employed taking two vital factors into consideration, namely, the codons that favored highly abundant E. coli proteins and, more importantly the highly specific restriction sites of the actual gene. The cassette included the 99-residue HIV protease gene, followed by a stop codon with 10 upstream residues. This was then ligated to a plasmid to give rise to a pUC12-HIV-PR plasmid, which contains the entire synthetic HIV-1 protease gene. Following this, an expression vector pET-HIV-PR was created to synthesize a 14-kDa precursor, which is the fusion of 16 amino acid residues from the expression vector to 109 residues of the HIV sequence [18]. The bacterium was transformed with this vector, and the enzyme was expressed.
Using this strategy, a plasmid encoding for HIV-1 protease isolated from HIV-1 subtype C (CSA) specifically was generated by Mosebi et al. [8] employing a site-directed mutagenesis approach to transform E. coli BL21 (DE3) pLyss cells. The coding region of the protease in the transgenic strain was confirmed by DNA sequencing. The protease was overexpressed as inclusion bodies and purified to yield a product that had a monomeric size of 11 kDa when resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and an apparent oligomeric molecular mass of 22 kDa [8]. In this study, it was shown that the protease isolated from HIV-1 subtype C catalytic activity did not differ from its subtype B wild type. However, it was established that it was more resistant to commercially available anti-protease drugs such as Saquinavir, Ritonavir, Indinavir, and Nelfinavir. This confirmed that mutations in the different subtypes created multi-drug resistance. It is therefore imperative that further drug discovery be designed on the subtype-specific basis.
This communication describes the optimization of the above method originally derived by the Protein Structure-Function Research Unit (University of the Witwatersrand, Johannesburg, South Africa). To this end, we investigated growth medium composition for the sustenance of the transgenic E. coli strain as an option for increasing HIV protease enzyme production [19, 20].
2. MATERIALS AND METHODS
2.1. Materials
All chemicals, solvents, and media were purchased from Merck (Pty) Ltd, South Africa, unless otherwise stated. Antibiotics were purchased from Melford Laboratories Ltd, South Africa.
2.1.1. Strains
E. coli BL21 (DE3) pLyss cells containing the plasmid encoding the HIV-1 protease from HIV-1 subtype C (HIV-1 CSA) (containing the mutation Q7K designed to reduce the hypersensitive autolytic site) were a kind gift from Professor Y. Sayed of the Protein Structure-Function Research Unit (University of the Witwatersrand, Johannesburg, South Africa).
2.1.2. Media
Cells were grown in two different types of media during expression in order to evaluate which medium results in quicker culturing time and higher protein yields. The first medium used was Luria Bertani (LB) broth containing 1% (wt/vol) Trypton, 0.5% (wt/vol) yeast extract and 0.5% (wt/vol) NaCl, supplemented with 100 μg/mL of ampicillin and 35 μg/mL of chloramphenicol. The second medium used was a chemically defined medium (Table 1) supplemented with 100 μg/mL of ampicillin,35 μg/mL of chloramphenicol [13].
| Component | Quantity |
|---|---|
| ATM4S solution | |
| Ammonium sulphate (NH4)2SO4 | 4 g |
| Sodium dihydrogen phosphate (NaH2PO4.H20) | 5.2 g |
| Di-potassium hydrogen phosphate (K2HPO4) | 12.5 g |
| Tri-sodium citrate (Na3-citrate.2H2O) | 0.66 g |
| Make up with deionised water (18 mΩ) to | 295 mL |
| Glucose solution | |
| Glucose Monohydrate | 1 g |
| Magnesium sulphate heptahydrate | 0.050 g |
| Make up with deionised water (18 mΩ) to | 250 mL |
| Yeast extract solution | |
| Yeast Extract | 10 g |
| Make up with deionised water (18 mΩ) to | 150 mL |
| Trace metal stock solution | |
| Na-EDTA | 20.1 g/L |
| FeCl3.6H2O | 16.7 g/L |
| ZnCl2 | 0.13 g/L |
| CoCl2.6H2O | 0.18 g/L |
| NaMoO4.2H2O | 0.13 g/L |
| CaCl2 | 0.50 g/L |
| CuSO4.5H2O | 0.16 g/L |
| H3BO3 | 0.28 g/L |
| MnCl2.2H2O | 0.11 g/L |
| Remove from stock solution | 2 ml |
*Combine 295,250, 150 and 2 ml of respective solutions and bring up to 1 L with deionised water (18 mΩ).
2.2. Overexpression and Extraction of HIV-PR
The E. coli cells harboring the plasmid DNA were grown in LB broth medium supplemented with antibiotics (100 μg/mL of ampicillin and 35 μg/mL of chloramphenicol). The overnight culture was diluted 100-fold by adding 10 mL to 990 mL to fresh LB broth supplemented with ampicillin (100 µg/mL) and chloramphenicol (35 µg/mL). Induction of the overexpression of the HIV-PR CSA was achieved by adding isopropyl β-D thiogalactoside [IPTG] (Sigma Aldrich, Germany) to a final concentration of 0.4 mM, and induction was allowed to proceed for a further 4 hours. Once the incubation period for overexpression of the protease was complete, the cells were collected in pre-sterilized centrifuge tubes and pelleted via centrifugation at 5000 x g for 12 minutes using an Avanti centrifuge J-26XPI (Beckman Coulter, USA) at 4 °C. The pellet was re-suspended in 50 mL of cold extraction buffer [10 mM Tris, 2 mM EDTA and 1 mM phenylmethylsulphonyl fluoride [PMSF] (added fresh before use) pH 8.0]. Both pellet and supernatant were analysed by SDS-PAGE even though the protease was within inclusion bodies that was present in the pellet. The volume was brought up to 100 mL with extraction buffer and treated with 10 mM magnesium chloride followed by homogenization (Bio-Gen PRO200 homogenizer) until a uniform solution was obtained. Maintaining a 4 ᵒC environment, DNAse1 (Thermo Scientific, South Africa) at a concentration of 5 µg/mL was added whilst stirring to remove all traces of DNA. Once the viscosity of the mixture was decreased, 20 mL aliquots of the solution were transferred to sterile Falcon tubes and exposed to vigorous sonication at 4 ºC using a Qsonica (Q125) probe sonicator (USA). Cells were then pelleted by centrifugation at 25000 x g for 30 minutes at 4 ᵒC and were re-suspended in extraction buffer containing 1% (v/v) Triton X-100. The homogenization and centrifugation steps were repeated, and the pellet was re-suspended first in ice-cold extraction buffer and then in solubilization buffer (10 mM Tris-HCL, 8 M urea, 2 mM DTT: pH 8) at room temperature. The supernatant was then collected after final centrifugation for further purification.
2.3. Purification and Renaturation of HIV-PR
HIV-PR was purified as described previously by Pawar et al. [21] with modifications. Briefly, the HIV-PR in the supernatant was purified by passing it through an anion exchange (DEAE) column (BIO-RAD laboratories, USA) previously regenerated with ten volumes of NaCl and then equilibrated with an equal volume of solubilization buffer at room temperature. Fractions (5 mL) were collected and subjected to the Bradford assay (Sigma Aldrich, Germany) to determine the presence of the protease. Fractions containing the protease were pooled and formic acid (Sigma Aldrich, Germany) was added to a final concentration of 25 mM, resulting in the reduction of the pH to approximately 3.0. The chromatography was monitored by UV detection at 280 nm using an automated microplate reader (Synergy HT, BioTek Instruments, South Africa) [20]. The fractions with the highest intensity were pooled for further protein purification. Fractions were stored at 4 ᵒC to precipitate protein contaminants. Following overnight incubation, the precipitated contaminants were removed by centrifugation at 15 000 x g for 30 minutes at 4 °C. The HIV-1 protease was refolded by dialysis against 10 mM sodium acetate, 1 mM NaCl and 1 mM DTT, pH 5.0 at 4 OC. Subsequently, the protease was dialyzed into storage buffer containing 10 mM sodium acetate, 1 mM NaCl and 1 mM DTT, pH 5.0. The folded protease was concentrated to a final volume of w5 mL and stored at -20 oC. The concentration of the protease was determined by using Beer-Lamberts law at the standard protein absorbance wavelength of 280 nm using an AnalytikJena Specord 210 spectrophotometer. The HIV protease expression was confirmed by using SDS-PAGE (Section 2.4). The specific activities of purified renatured HIV PR were determined as described in Section 2.5.
2.4. Sodium Dodecyl Sulfate-polyacrylamide Gel Electrophoresis (SDS-PAGE)
Samples (20 µL) were electrophoresed in a 15% SDS-PAGE system using Mini-Protean TGXTM (BIO-RAD laboratories (USA) [21]. The gel was stained using PageBlueTM protein staining solution containing Coomassie brilliant blue G-250 dye and de-stained in 40% methanol and 10% acetic acid mixture. The gels were then captured using a Syngene gel documentation system (Vacutec, South Africa).
2.5. Assay of HIV-PR CSA Protease Activity
The specific activity of HIV-PR CSA was determined using a chromogenic peptide substrate Lys-Ala-Val-Nle-p-nitro-Phe-Glu-AlaNle-NH2, which mimics the conserved KARVL/AEAM cleavage site between the capsid protein and nucleocapsid (CA-p2) within the Gag poly-protein (chromogenic peptide was a kind gift from Dr. M. Makatini (School of Chemistry, University of KwaZulu-Natal, Durban, South Africa) [22]. The reaction mixture consisted of 50 μM of both substrate and protein (within range of 100-200nM) and 50 mM sodium acetate buffer (pH 5, with 0.1 M sodium chloride), at a final volume of 120 μL. Protease hydrolytic activity was measured by monitoring the relative decrease in absorbance at 300 nm using an AnalytikJena Specord210 spectrophotometer over a time period of 3 minutes.
3. RESULTS
3.1. Media Optimization
3.1.1. Growth Curve Analysis
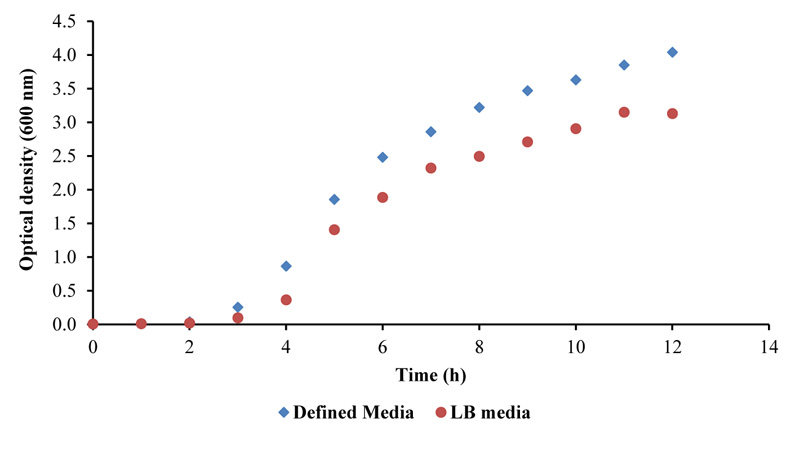
In this study, two different types of media for the overexpression of the protease were evaluated. LB and a specialized chemically defined media (Table 1), both supplemented with antibiotics, were used to determine which media resulted in higher cell density, thus potentially leading to a greater yield of overexpressed protein product [13]. A standard growth curve of the bacteria was analyzed using the optical density at 600 nm as a measurement of cell density.
3.2. Optimization of HIV-PR Production Using Different Media
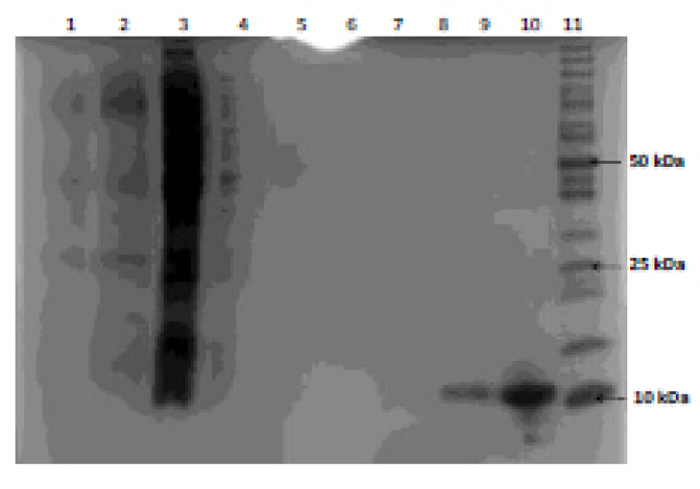
Recombinant E. coli was grown in the two different types of media and subsequent protein expression, extraction and purification were performed. Protein determination using SDS-PAGE (Fig. 2) and protein concentration were evaluated using spectrophotometric analysis (Table 1). The supernatant was collected during centrifugation steps in the extraction phase of the protease so that the loss of enzyme could be traced to a specific part of the method in the event of unsuccessful protease purification. Filtrate during the concentration steps in the above method was also analyzed for traces of lost protease.
| Purification steps | Protein Concentration (µM) | |
|---|---|---|
| Medium | ||
| Luria Bertani (LB) | 22.1 | |
| Chemically defined | 44.2 | |
| DEAE eluent fraction | ||
| “peak” | 44.2 | |
| “tail” | 10.5 | |
| SDS-PAGE purified sample | ||
| Lane 2 | 68.2 | |
| Lane 3 | 8.4 | |
| Lane 4 | 62.8 | |
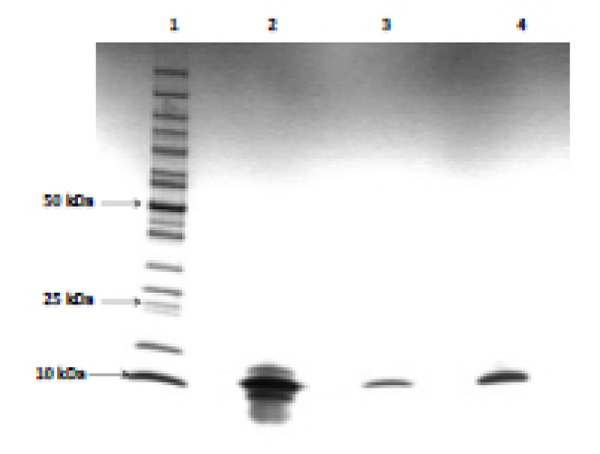
As shown in Fig. (2), HIV PR was successfully expressed as evident by a band size confirmation of 11 kDa (lane 9 and lane 10) and the absence of protease in the filtrate and supernatant confirms (lanes 4 to 8) that protease purification was successful. The abundance of protein present in lanes 2 and 3 is evidence of cell proteins extracted during the extraction of the inclusion body. Filtrate analysis indicated that the concentration of the protease was achieved. Furthermore, protease purified from E. coli grown in chemically defined medium (Lane 10) resulted in twice the amount of protease produced compared to E. coli grown in LB medium (Lane 9); which was further confirmed by protein determination (Table 2).
3.3. Optimization of the HIV-PR Purification
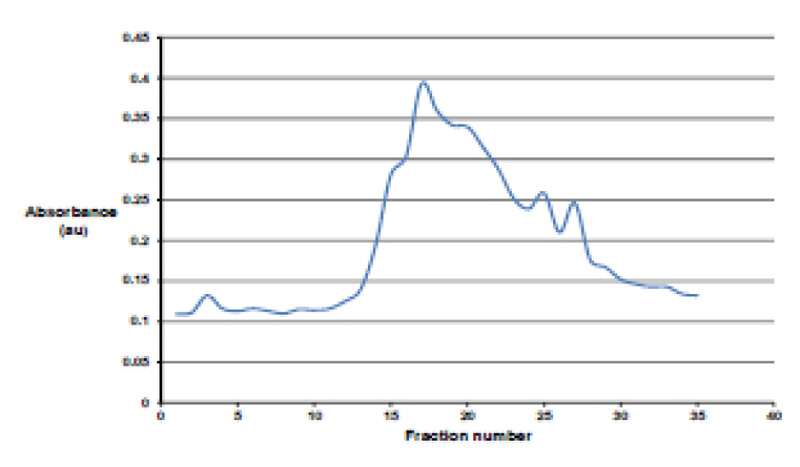
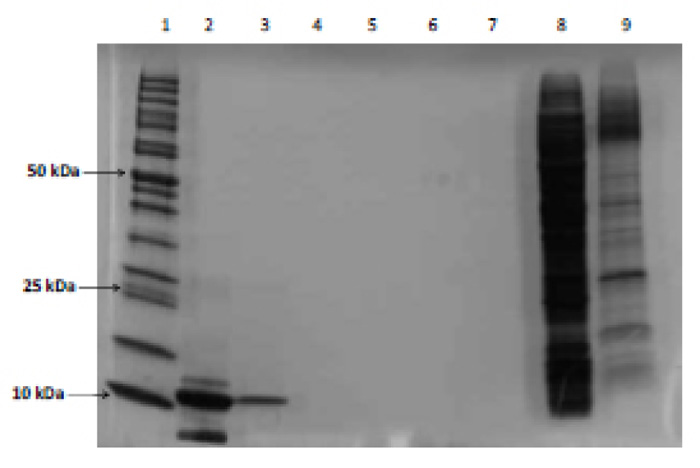
All fractions collected from the DEAE column were analyzed spectrophotometrically at 280 nm to determine which fractions contained the protease (Fig. 3). The protease purification was achieved using fractions 15-25. This was further divided into two groups, namely “tail,” which utilized fractions 15-18 and 21-25, and “peak”, which contained fractions 18-21. Fig. (4) shows an impure protease with multiple bands present in the “tail” fraction.
Comparison of the results (Fig. 5) show that the UV spectrophotometric analysis of the pooled fractions eluted by the column was not reliable due to multiple band formation and loss of protease due to their presence in the “tail” fraction. However, analysis of pooled fractions by Bradford assay (Lane 4) resulted in one band indicating a pure protease. Therefore, the final method for optimization included the use of the chemically defined media and fraction testing using the Bradford assay.
3.4. Protease conformation
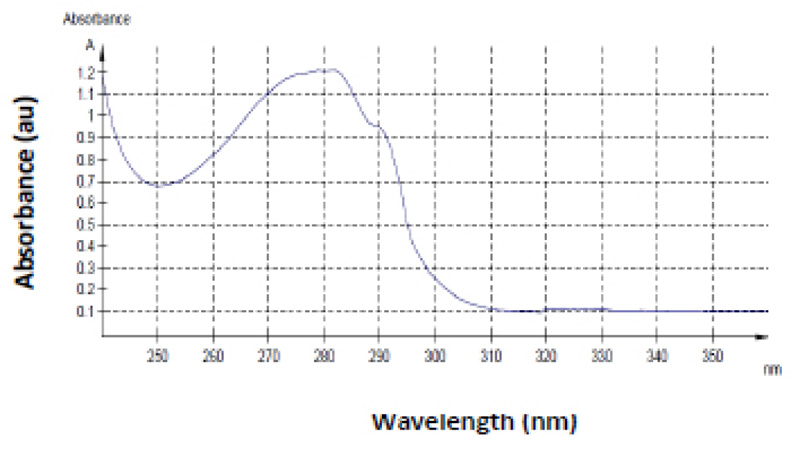
Besides size confirmation, further verification of the purified product was performed by UV spectrum analysis of the purified product. This resulted in the expected spectral data (Fig. 6). The amino acids that makeup proteins absorb ultraviolet light very strongly and are commonly identified at 280 nm. Mostly the aromatic residues, tryptophan, tyrosine, and cysteine, are responsible for absorbance at this wavelength and these are found in abundance in the 99 amino acid HIV-1 protease [11, 23]. The spectral data revealed a peak at 280 nm, confirming the presence of a pure protein. For reasons unexplained, the HIV protease spectral scan has a characteristic “shoulder” approximately at 290 nm as seen in Fig. (6), this can be an additional confirmation for a pure protease. This result correlates to data represented in studies of this nature [24].
3.5. Enzyme Activity
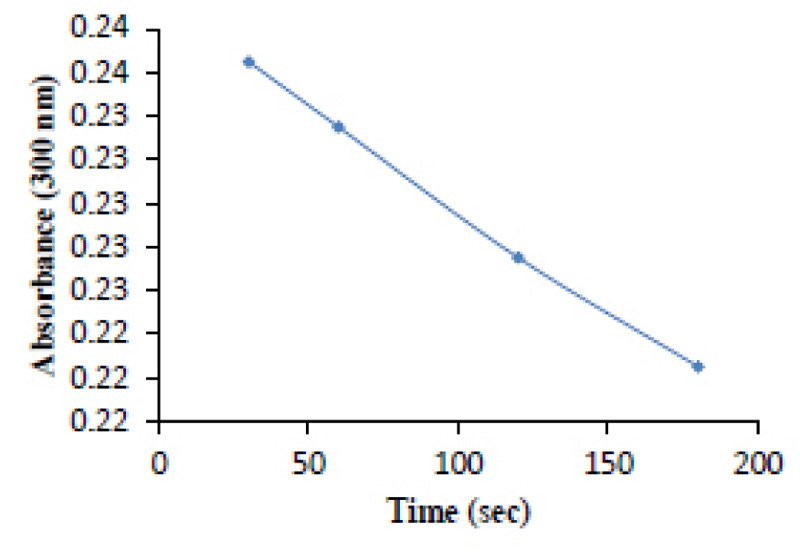
The purified protease sample was tested for catalytic activity by the use of a chromogenic substrate (Section 2.5). The hydrolysis of the substrate is an indication of the HIV-PR CSA’s ability to specifically bind to the substrate as well as cleave it appropriately. As time increased, the absorbance of the substrate decreased, which indicates substrate hydrolysis (Fig. 7).
*The experiment was carried out at 20 °C in 10 mM sodium acetate buffer, 0.1 M sodium chloride, pH 5.0.
4. DISCUSSION
Growth potential of E. coli harbouring transgenes for the expression of recombinant proteins is an area of interest for many reasons, particularly the impact of high cell density cultivation on recombinant proteins [25-27]. Manipulating the feeding strategy of these organisms alters their protein output in various ways. The consensus of the nutrient control is that a decrease in glucose content eliminates the probability of over-feeding and excessive by-product formation, which may inhibit growth. It has been previously established that the depletion of certain key amino acids encourages increased recombinant protein expression. This is due to the fact that these proteins are not composed of the essential amino acids that are required by the indigenous E. coli proteins [13, 28-30].
Studies indicate that recombinant bacteria undergo metabolic stress when expressing transgenes, thereby leading to low cell density and low protein concentration. However, it has been proven that using a chemically defined media that contains certain additives of the tricarboxylic acid (TCA) cycle, relieves the burden of metabolic stress caused on the bacterium, therefore, enhancing optimal heterologous protein synthesis [13, 29, 30].
In a study conducted by Ramcahran et al. [13], recombinant E. coli was used to express the protease xylanase using additives from the TCA cycle to recreate the effects of metabolic stress. This study applies the same principle by adding TCA intermediates such as citrate to the growth medium (Table 1) to enhance the generation of HIV-PR CSA. From the results, using a similar chemically defined medium promoted greater cell biomass, which generated a higher protease concentration (Figs. 1 and 2, Table 1). The growth curve employed in this study was also used to determine the point in the growth phase where induction of expression is required. The point of induction of expression is of great importance because the inducer, if used untimeously, would significantly contribute to metabolic stress of the bacterium and ultimately low heterologous protein expression. The best time for induction is the mid-log phase, which was established to be after 4.5 hours (Fig. 1). The optimization strategy employing chemically defined medium clearly indicates a higher protease yield.
To optimize HIV protease, the yield of all fractions eluted from the cation exchange column was monitored using both the Bradford and spectrophotometric methods. However, pooling of selected eluted fractions using spectrophotometric analysis yielded an impure protease as the end product (Figs. 4 and 5). Although the expected protease band size was evident, multiple band formation was also evident (Figs. 4 and 5). Furthermore, the fractions consisted of the “tail” generated small quantities of protease. Conversely, when the monitoring of fractions was performed using the Bradford assay, a pure protease product was achieved (Figs. 5). Based on the above results, it is proposed that the Bradford method be employed as a preferred method to pool fractions to ensure greater and more purified protease preparations.
These findings are supported by the proposed method by Mosebi et al. [8], and correlate with data obtained from Naicker et al. and Maputsoe et al. [31, 32]. HIV-1 PR was clear by visualizing a single 11 kDa band upon SDS-PAGE analysis (Fig. 5). Ultra-violet spectrophotometric analysis of the renatured and purified protease (Fig. 6) confirmed the presence of the protease since the unique finger print “shoulder” between 285 and 295 nm is a characteristic that is indicative of the HIV-1 protease [32, 33].
HIV-1 protease catalyzes the hydrolysis of peptide bonds with high sequence selectivity and catalytic proficiency. The substrate was designed to accommodate these specific amino acids and is, therefore, a suitable candidate for selective binding. Therefore, further evidence of a pure and active HIV-PR CSA was exhibited in an enzyme-specific reaction, and it was revealed that the specific activity of the enzyme was a positive enzyme reaction. Since HIV protease cleaves the specific sequence of the chromogenic peptide [19, 34, 35], thus decreasing its absorbance, it is safe to accept that the enzyme has the efficient ability to selectively cleave the substrate (Fig. 7).
CONCLUSION
In this study, we reported an optimized method for HIV-1 protease derived from HIV-1 subtype C production. This was achieved using a known method to isolate and purify the enzyme and with the use of a specialized feeding strategy. , A combination of these optimisation optimization strategies yielded an 11 kDa pure protein precursor from the overexpressed gene of interest. The protease generated also displayed anticipated catalytic activity. It was established that the growth of the transgenic strain in a chemically defined media yielded a 100% increase in HIV-1 PR production. The HIV-1 protease produced in this study was subsequently used to chemically test the enzyme inhibition capabilities of the novel cage and sugar-based peptide HIV protease inhibitors that were synthesized at this institute. Further work on the structure, dynamics, and modification, etc., of HIV-1 protease isolated from HIV-1 subtype C will be undertaken due to the optimization strategy for HIV-1 protease production.
AUTHORS’ CONTRIBUTIONS
Uraisha Ramlucken performed the experiments and data analysis. Patrick Govender designed the experiments and reviewed the manuscript. Krishna Suresh Babu Naidu prepared the manuscript, data analysis and edited manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
All patients participated on a voluntary basis and gave their informed consent.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is available in the [Figshare.com] at [https://doi.org/10.6084/m9.figshare.13302248.v1], reference number [13302248]”.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
FUNDING
This study was made possible through financial support from the National Research Foundation.
ACKNOWLEDGEMENTS
The authors would like to thank the National Research Foundation for funding and the University of KwaZulu-Natal for providing research facilities.

