All published articles of this journal are available on ScienceDirect.

Bioinformatic Analysis of the Human Recombinant Iduronate 2-Sulfate Sulfatase
Authors Info & Affiliations
Abstract
Mucopolysaccharidosis type II is a human recessive disease linked to the X chromosome caused by deficiency of lysosomal enzyme Iduronate 2-Sulfate Sulfatase (IDS), which leads to accumulation of glycosaminoglycans in tissues and organs. The human enzyme has been expressed in Escherichia coli and Pichia pastoris in attempt to develop more successful expression systems that allow the production of recombinant IDS for Enzyme Replacement Therapy (ERT). However, the preservation of native signal peptide in the sequence has caused conflicts in processing and recognition in the past, which led to problems in expression and enzyme activity. With the main object being the improvement of the expression system, we eliminate the native signal peptide of human recombinant IDS. The resulting sequence showed two modified codons, thus, our study aimed to analyze computationally the nucleotide sequence of the IDSnh without signal peptide in order to determine the 3D structure and other biochemical properties to compare them with the native human IDS (IDSnh). Results showed that there are no significant differences between both molecules in spite of the two-codon modifications detected in the recombinant DNA sequence.
INTRODUCTION
Mucopolysaccharidosis type II (MPS II), also known as Hunter syndrome, is an inherited lysosomal storage disease that is recessively transmitted and X-linked. It is caused by an IDS enzyme deficiency, (EC 3.1.6.13), which generates tissue accumulation of mucopolysaccharides, heparan and dermatan sulfate. This results in the urinary excretion of these very same molecules; fact that has been used for the diagnosis of the disease [1].
Like in other Inborn Errors of Metabolism (IEMs), the cause of this disease derives from enzyme coding gene and results in a condition that is untreatable by conventional methods [1]. Historically, MPS II management has been done through the correction of clinical manifestations by means of supportive therapy. One of the alternatives recently proposed for the treatment of lysosomal storage diseases is ERT, which has shown to be an alternative to reverse the visceral complications in diseases such as Hunter syndrome [2, 3].
In this respect, the search for new therapeutic solutions has brought about the ability to provide patients with active IDS enzyme by ERT. Elaprase™, a recombinant enzyme, which is produced in a human cell line [2]. ERT has also shown to be effective in other metabolic disorders such as Gaucher, Fabry, Hurler, Maroteaux-Lamy, and Pompe disease [1]. Nonetheless, keeping in mind that ERTs are costly for third world countries, it is necessary to further develop recombinant protein expression systems [4] with more competitive cost models than those of human cell lines are, in order to finally be able to provide benefits to the patients and the domestic industry. Thus, the use of lower costs expression systems, such as the bacterium E. coli [5, 6] and the yeast P. pastoris [7, 8], have opened up new horizons that allow the continuation of exploring strategies for treatment of MPS II among other IEMs.
The present study aimed to computationally analyze the nucleotide sequence of the IDSnh without signal peptide (IDSwsp) subcloned into pGEM-T Easy which was designated as pGEM-T Easy-IDSwsp and whose subsequent objective was to clone the IDSwsp in the E. coli K12 strain GI724 system, using the pLEX-IDSwsp genetic construction [9].
MATERIALS AND METHODS
IDSwsp Sequencing
The subcloned gene (pGEM-T Easy-IDSwsp) was sequenced at the Instituto Nacional de Salud (http://www.ins.gov.co) following the Sanger method [10]. The reaction was carried out in an ABI PRISM 377 automated sequencer using the BigDye Terminator v1.1 cycle sequencing reaction mixture. The oligonucleotides used for sequencing were those corresponding to the T7 and SP6 promoters, which flank the multiple cloning site of the pGEM-T Easy, as well as the IDS-F and IDS-R primers previously used to obtain the IDSwsp (Table 1).
Primers used to amplify the cDNA of the IDS without the sequence it encodes for the native signal peptide. IDS-F (28 nucleotides) contains an EcoRI recognition site (underlined). IDS-R (26 nucleotides), contains a Not I recognition site (underlined).
| Primer | Sequence 5´- 3´ | Number of nucleotides |
|---|---|---|
|
T7 |
TAATACGACTCACTATAGGG |
20 |
|
SP6 |
TATTTAGGTGACACTATAG |
19 |
|
IDS-F |
GGAATTCTCCGAAACGCAGGCCAACTCG |
28 |
|
IDS-R |
GGCGGCCGCACACTGAGGGATGTCTG |
26 |
Bioinformatics Analysis of the IDS Enzyme Without Native Signal Peptide
The access numbers used for IDS are AAA63197 (http://www.ncbi.nlm.nih.gov) in GenBank, 91046030 in Medline and 2122463 in PubMed. There, the IDS amino acid sequence was taken in the FASTA format needed for computational studies and obtained from the conceptual translation of the cDNA reported by Wilson [11, 12].
ProtScale of ExPASy (http://www.expasy.org) was used to determine accessibility, hydrophobicity, polarity, and flexibility profiles [13]. As the software recommends it, a basic nine amino acids window size was used for the ProtScale analyses. This ensures optimal coverage of the sequence. In the case of the hydrophobicity profile, the Kite and Doolittle algorithm was applied. Its scale considers values between -3.0 and 4.0 [14]. The Compute pI/MW software allowed calculating the isoelectric point (pI) and molecular weight (MW), [15, 16]. Potential phosphorylation sites were determined with NetPhos 2.0 [17]; those of O- and N-glycosylation were determined with NetOGlyc 3.1 [18, 19] and NetNGlyc 1.0 software (http://www.cbs.dtu.dk), [20] respectively.
The IDSnh and IDSwsp amino acid sequences were provided to the online Phyre 2.0 server through the Structural Bioinformatics Group, Imperial College London (http://www.sbg.bio.ic.ac.uk). Phyre is specialized in tertiary structure prediction software. It is based on remote homology modeling and fold recognition, which function through a profile-to-profile matching algorithm [21]. The template identified by Phyre for both proteins was a crystallographic structure with a resolution of 1.7 Å of a putative sulfatase from Bacteroides fragilis registered in the Protein Data Bank (PDB) (http://www.rcsb.org) with the access code 2QZU. The refinement and minimization of the models was performed using the UCSF CHIMERA software [22].
Geometry assessment was performed with the QMEAN server from Swiss Institute of Bioinformatics (SIB). QMEAN software, estimates the model structure quality by making an exhaustive comparison with the structures determined by crystallography or by the magnetic resonance deposited in the PDB. It uses two scoring functions: QMEANlocal and QMEANclust [23, 24]. To estimate the correct arrangement of the dihedral angles (ψ/Ф) a Ramachandran plot that ignores the glycine and proline residues was generated by the RAMPAGE server (http://mordred.bioc.cam.ac.uk). The RMSD analysis used to determine the structural homology came about by comparing the models obtained through the SuperPose 1.0 software [25]. The architecture of the models was analyzed with the Vadar 1.8 software [26], which calculates the average distance of the hydrogen bonds, dihedral angles, accessible surface area, and the models’ volumes.
RESULTS AND DISCUSSION
pGEM-T Easy-IDSwsp Sequencing
The IDSwsp nucleotide sequence obtained (Fig. 1A) was compared using the ClustalW2 program (http://www. ebi.ac.uk) with the sequence corresponding to variant 2 of the human IDS mRNA (GeneBank access code: NM_006123.4). The homology between the two sequences is shown in Fig. (1B). Two substitutions were observed in the consensus sequence. The first was noticed in nucleotide 1201 where a cytosine is replaced with a thymine, and the second in nucleotide 1256 where a thymine is replaced by a cytosine.
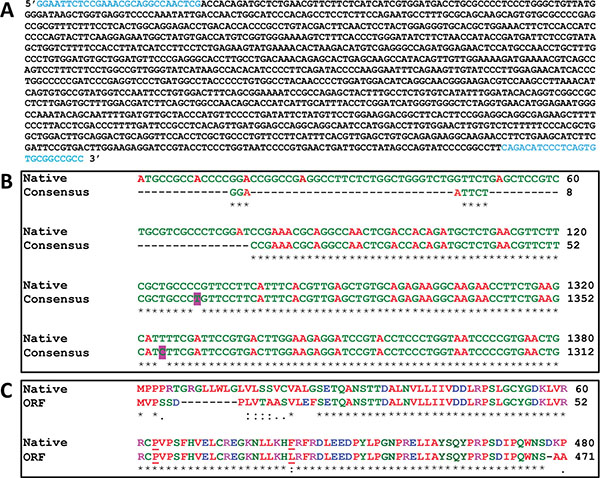
(A). IDSwsp nucleotide sequence. In blue, the sequences corresponding to the IDS-F and IDS-R primers are shown. (B), Alignment of the IDSwsp consensus and IDSnh nucleotide sequences. An observation of the homology between the two sequences, which shows that the IDS consensus starts right after the sequence that encodes for the peptide signal (first 75 nucleotides). Highlighted in pink two substitutions can be observed, one in nucleotide 1201 (C/T) and another in nucleotide 1256 (T/C). For the native sequence, the nucleotides between 121 and 1260 are omitted, while for the consensus sequence, the nucleotides between 53 and 1192 are omitted. (C). Alignment of the IDSnh (Native) and IDSwsp ORF amino acid sequences. Those underlined indicate the amino acids involved in substitutions, where the substitution C/T in the third nucleotide of codon 415 produces P, the same amino acid as that of the native sequence. The T/C substitution in the first nucleotide of codon 434 causes an exchange of F for L. For the native sequence, the amino acids between 61 and 420 are omitted, while for ORF the amino acids between 53 and 412 are omitted.
For this reason, in order to attain an IDS complete analysis that is anticipated to be expressed in different vectors sometime in the future, a translation of the IDSwsp Open Reading Frame (ORF) was made and then compared to the sequence of IDSnh amino acid residues. Translate software from the SIB (ExPASy) was used to accomplish this.
Fig. (1C) shows the alignment of the amino acid residue sequences of IDSnh and IDSwsp obtained in this work. Despite the two substitutions found the sequence corresponding to IDSwsp, shows a high degree of identity when compared to the IDSnh. The latter is due to the replacement of the third nucleotide in codon 415, which resulted in a synonymous substitution. In both cases, Proline was the amino acid residue produced. The substitution of the first nucleotide in codon 434 resulted in the exchange of phenylalanine (F) residue for leucine; protein functionality will not be compromised since both belong to the group of amino acids. However, taking into consideration that the F is an aromatic amino acid, there is the possibility that the structure be affected more by the size of amino acids than by the characteristics related to the polarity of these. Additionally, it is also important to consider changes that may generate additional amino acids, which would arise due to the characteristics featured in the multiple cloning sites of the expression vectors. For these reasons, it is essential to understand the implications that these changes could generate in the 3D conformation of the structure, before starting expression experiments in future work.
Bioinformatics Analysis of the Recombinant Enzyme IDSwsp
Currently, over thirty million unique protein sequences have been deposited in public databases, a number that continues to grow rapidly (http://www.ncbi.nlm.nih.gov). However, only about 100,000 protein structures have been determined experimentally (http://www.rcsb.org) despite the progress in research initiatives. In the specific case of human proteins, it should be highlighted that difficulties arise when attempting to purify them from their natural sources. This disparity between the number of sequences and structures has led researchers towards the use of computational methods for the prediction of protein structures based on amino acid sequences. Consequently, computer programs have become useful tools. These programs facilitate experimental designs by providing essential knowledge of aspects relating to structure, and therefore to molecule behavior as well. Hence, computer programs save energy, time, and money which otherwise would not be possible. The results obtained through the application of these tools, a thorough literature review, and theoretical analyses can be very useful. These results could contribute to the improvement of the expression and to the potential use of recombinant IDS in ERT for patients with Hunter syndrome.
Computational analyses were carried out at a primary structure level, and models were formulated for the secondary and third structures. These analyses were conducted in a comparative manner between the IDSnh and the IDSwsp sequence in order to determine the influence of the additional amino acids that are present at the amino and carboxyl terminus, and of the mutations that were found in the sequence.
Hydrophobicity profiles were obtained by applying the Kyte-Doolittle algorithm, which employs a scale that assigns hydrophobicity values for each amino acid with variations of -3.5 (glutamate) and 4.2 (valine). Meanwhile, polarity profiles were obtained by applying the Zimmerman algorithm, which employs a scale that assigns polarity values to each of the amino acids with variations of 0.0 (alanine) and 52 (arginine). For flexibility, the Bhaskaran scale was employed, which assigns flexibility values for each of the amino acids with variations of 0.30 y 0.54. Ultimately, for percentage accessibility the scale used assigned accessibility percentage to each of the amino acids with variations of 0.3 (lysine) and 0.9 (cysteine).
In general, the profiles obtained for IDSnh and for IDSwsp show low levels of hydrophobicity and high levels of polarity. This suggests that the molecules are hardly hydrophobic and predominantly polar. In addition, the high flexibility and accessibility values obtained indicate that the molecules are flexible and possess highly accessible regions (Fig. 2A).
In large part of the IDSnh and the IDSwsp molecules, the results obtained from the hydrophobicity profile prediction are consistent with the high percentage of exposed areas in the tertiary structure and with its lysosomal enzyme nature, which is in constant interaction with polar phases. This enzyme requires being flexible in order to carry out its function; this property is explained by the high flexibility of the molecules. In addition, the prediction of the accessibility profiles suggests that IDSnh and IDSwsp are molecules with a high percentage of highly accessible areas, which is consistent with the high percentage of tertiary structure in random coil formation.
When comparing both molecules for all the analyzed profiles, there is no observation of any variations in the behavior of the IDSwsp molecule with respect to the IDSnh molecule that would suggest an abrupt alteration caused by mutations present in the sequence. However, it should be pointed out that there are 17 additional amino acids present in the IDSwsp amino terminus. Without remarkably altering the overall behavior of each profile, these amino acids generate some changes in the profiles at the beginning of the sequence.
Computational prediction values for the pI and MW were 5.15 and 58.479 kDa for the IDSnh, and 5.33 and 53.473 kDa for the IDSwsp. Experimentally, similar values have been determined in terms of pI. However, MWs between 43 and 49 kDa have been reported for the mature protein in different cell types [12, 27, 28] when applying other methods to determine MW. This difference is possibly due to the mature human IDS forms that have lost a fragment of 18 kDa in the process, which determines that the mature sequence extends up to the residue 456 in IDSnh and up to 448 in IDSwsp.
As for potential phosphorylation sites, 26 were found for IDSnh and 25 for IDSwsp. However, (as shown in Fig. 2B) IDSwsp presents an additional potential phosphorylation site of a serine amino terminus and lacks in the carboxyl terminus of two serine and tyrosine potential phosphorylation sites.
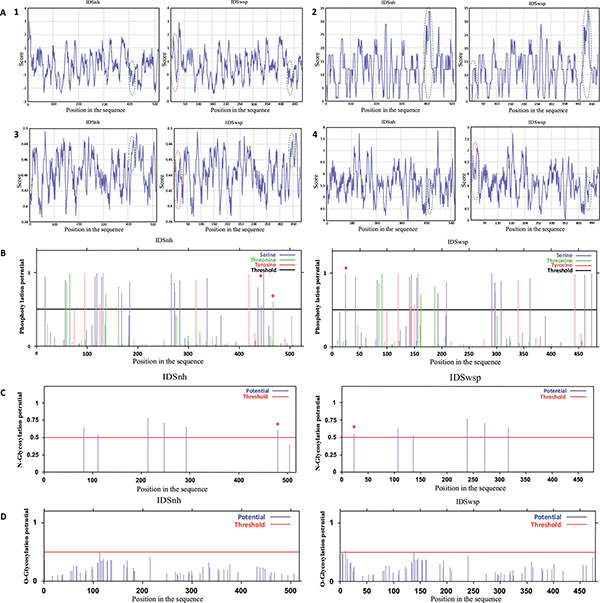
(A). Profiles obtained in the analysis of the primary structures of IDSnh and IDSwsp 1. Hydrophobicity, 2. Polarity, 3. Flexibility, and 4. Accessibility of IDSnh and IDSwsp. Highlighted in red are the areas of the amino terminus zones of the IDSwsp; presented as additional amino acids due to the expression vector sequence. Green represents the comparison between the IDSnh areas corresponding to the mutations found in the IDSwsp. (B). Potential phosphorylation sites of IDSnh and IDSwsp using the NetPhos 2.0 * program.(C). Potential sites of N-glycosylation of the IDSnh and IDSwsp using the NetNGlyc 1.0 * program.(D). Potential sites for O-glycosylation of the IDSnh and IDSwsp, using the NetO-Glyc 3.1 * program. The differences are marked with an asterisk (*).
It is important to emphasize that phosphorylation could change the charge of a protein and could cause a conformational change of the molecule. These effects can significantly alter the binding of the ligand to the protein and lead to an increase or decrease of its activity. However, it should be taken into account the closeness or relation of the amino acid to the active site of the enzyme, as predictions of potential phosphorylation sites are conducted under the analysis of the primary structure and provide data that indicate that it is possible for an event to occur. Yet, factors corresponding to the final 3D configuration that is generated from the primary sequence are not taken into account. In other words, whether phosphorylation finally occurs or not is also due to related factors, for example, the location of such amino acid in the folded molecule. Thus, if an amino acid is found facing the interior of the molecule, it would be less susceptible to a phosphorylation in compare to an amino acid that appears exposed in the structure, because the contact of the protein kinases involved in the phosphorylation process would be favored.
As for glycosylations, they could be mainly of two types, N- and O-glycosylations. Because of its lysosomal nature human IDS undergoes a series of posttranslational modifications, amongst which the glycosylation process is one of the most important. The computational component indicates that in the IDSnh peptide precursor, seven potential sites for glycosylation of asparagine residues (N-glycosylation) exist; the last two are lost when a fragment of 18 kDa is removed. Four of these sites have a 65 to 80% probability of glycosylating, two have a 50 to 60% probability, and one has a probability of less than 50%. Regarding the potential N-glycosylation sites for IDSwsp, an additional sequence is present at the start site; however, the probability of this one taking place fails to exceed 55% (Fig. 2C), suggesting that the variation of the IDSwsp N-glycosylation sites in regard to the IDSnh presents no major differences. Lastly, of the IDSnh and IDSwsp potential O-glycosylation sites, neither has a probability greater than 50%, thus they are not considered potential glycosylation sites (Fig. 2D).
Prediction by homologous modeling offers the possibility of creating real approximations to the 3D structure of the protein of interest. It could also be applied to experimental designs and to the explanation of results. In general, the first step in these prediction processes is carried out with energy minimization algorithms. The algorithms report one of the models in one of the possible energy minimums of the surface, which is the full resolution to the functional equations of potential energy. A geometry optimized in a potential energy minimum with only one possible structure was obtained for this work. Sets of differential equations were solving by following the principle of stability, as is the case of the potential energy of a molecule. It is understood as if there are two stable solutions, which represent geometries for the proteins in each case, they must be very similar; otherwise, they would not meet the criteria of asymptotic stability suggested by Lyapunov [29].
It has been established that homologous proteins evolved from a common ancestor, retain their function through the biological scale, and adopt similar 3D structures. These proteins generally have sequences similar by about 20%, whereas analogous proteins reach up to about a 10% similarity [21, 30, 31]. Since similar sequences decrease in analogous and distant homologous proteins, the recognition of homologies and similarities is important in order to obtain an approximation of the 3D structure. The alignments of the IDSnh and the IDSwsp with the putative sulfatase of B. fragilis obtained in Phyre showed identities of 21% and 18% respectively. These values are within the ranges established for considering that a prediction has a high probability of occurring (Figs. 1, 2, 3A).
Models generated by the Phyre server (Figs. 3, 3A, 4) presented reliability prediction values higher than 90%, in 95% of the residues modeled for IDSnh and in 96% of those modeled for IDSwsp. This indicates that the structures obtained largely reflect the homologous models experimentally obtained, and therefore these structures are highly likely to occur in reality.
The validation of the 3D models obtained in the stereochemistry evaluation, indicates that the interatomic distances and torsion angles of peptide bonds are in allowed regions in the case of most amino acids (Fig. 3B). The scores obtained from QMEAN-Z correspond to the extent of the absolute quality of the models in comparison to models obtained experimentally, where models of low quality are expected to exhibit highly negative values. The results show values of -3.03 for IDSnh and of -3.65 for IDSwsp. These values are appropriate and could reflect the biological reality of the studied molecules.
When comparing IDSwsp structure to IDSnh, RMSD values of 2.12 Å were obtained. This means that the differences between the two models are not significant, and the differences are located random coil regions. Therefore, there are possibilities of expressing an active IDSwsp enzyme with a proper folding, or at least with one similar to the folding present in IDSnh (Fig. 3C).
Despite the limiting factors of computer models, it is important to emphasize that these provide the only feasible alternative for creating functional and structural predictions when the purified protein is not available in sufficient quantities to determine its structure experimentally. Furthermore, the predictions are very useful when designing the experimental processes, and in some cases, high correlation values have been found when making comparisons between theoretical predictions and experimental data. It should be noted that the structures derived from X-ray analysis and nuclear magnetic resonance also present limitations that so far have remained unresolved. Therefore, the information obtained computationally will serve as a guide, for designs of future experiments in attempt to improve the levels of enzyme expression and purification.
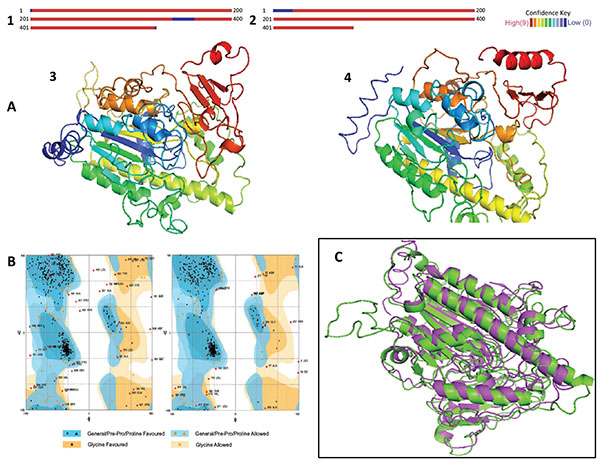
(A). Results of homology modeling and 3D models.1. IDSnh. 2. IDSwsp. The red areas indicate that the confidence level of the model is high, while blue colors indicate low confidence levels. 3 and 4. The models represent the 3D configuration of the IDSnh and the IDSwsp, respectively. The variations observed relate mainly to the additional amino acids in the amino and carboxyl terminus present in the IDSwsp. (B). Representation of the Ramachandran Plot, obtained for the IDSnh and the IDSwsp, respectively. It indicates whether the interatomic distances and torsion angles of the peptide bonds are in allowed regions in the majority of amino acids. (C). Superposition of the IDSnh and the IDSwsp. Green indicates the structure corresponding to IDSnh and purple indicates the structure corresponding to IDSwsp. The RMSD obtained was of 2.12 Å.
CONCLUSION
For several years, our research groups have directed a great part of the efforts in order to generate knowledge and to produce the recombinant enzyme [4, 8, 32, 33] for implementing ERT protocols for mucopolysaccharidosis, including Hunter syndrome. In this work, a computational model with a 3D structure of the IDSwsp was proposed. This exhibited 30% helix, 24% folded sheet, and 45% coil. According to hydrophobicity, polarity, flexibility, and accessibility profiles, it was determined that IDSwsp is hardly a hydrophobic molecule, but it is predominantly polar, flexible, and has high accessibility regions. The comparison of the models IDSwsp and IDSnh, indicates that it is very likely to obtain an active enzyme in future protein expression experiments, as in the present work, being not significant the differences found between the two molecules. Finally, our purpose in the long term is to produce recombinant proteins that could be use in the treatment of such diseases. Being necessary a consistent production of complex proteins like hrIDS, glycosylated (in P. pastoris) or not glycosylated (in E. coli); since Hunter Syndrome is considered an orphan disease and in most cases only supportive therapy is applied, where many of the Latin American health systems do not cover the elevated costs of such therapies.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This research was supported and funded by the Genetics and Biochemistry Laboratory and the Master’s Program of Biomedical Sciences of the Universidad del Quindío (Armenia, Colombia). Authors thank Alejandra González for the English editing of the manuscript.

