All published articles of this journal are available on ScienceDirect.

Molecular Identification of Nontuberculous Mycobacteria in Humans in Zimbabwe Using 16S Ribosequencing
Authors Info & Affiliations
Abstract
Background:
Several nontuberculous mycobacteria (NTM) were previously isolated from diverse environments such as water, soil, sewage, food and animals. Some of these NTM are now known to be opportunistic pathogens of humans.
Objective:
The main purpose of the study was to identify NTM isolates stored at the National Microbiology Reference Laboratory (NMRL) and were previously isolated from humans during a national tuberculosis (TB) survey.
Methods:
Pure NTM cultures already isolated from human sputum samples during the national TB survey were retrieved from the NMRL and used for this study. DNA was extracted from the samples and 16S ribosomal RNA gene amplified by polymerase chain reaction. The amplicons were sequenced and bioinformatics tools were used to identify the NTM species.
Results:
Out of total of 963 NTM isolates stored at the NMRL, 81 were retrieved for speciation. Forty isolates (49.4%) were found to belong to Mycobacterium avium-intracellulare complex (MAC) species. The other 41 isolates (50.6%) were identified as M. lentiflavum (6.2%), M. terrae complex (4.9%), M. paraense (4.9%), M. kansasii (3.7%), M. moriokaense (3.7%), M. asiaticum (2.5%), M. novocastrense (2.5%), M. brasiliensis (2.5%), M. elephantis (2.5%), M. paraffinicum (1.2%), M. bohemicum (1.2%), M. manitobense (1.2%), M. intermedium (1.2%), M. tuberculosis complex (1.2%), M. parakoreense (1.2%), M. florentinum (1.2%), M. litorale (1.2%), M. fluoranthenivorans (1.2%), M. sherrisii (1.2%), M. fortuitum (1.2%) and M septicum (1.2%). Two isolates (2.5%) could not be identified, but were closely related to M. montefiorense and M. phlei respectively. Interestingly, the MAC species were the commonest NTM during the survey.
Conclusion:
The study emphasizes the importance of identifying species of NTM in Zimbabwe. Future studies need to ascertain their true diversity and clinical relevance.
1. INTRODUCTION
Nontuberculous mycobacteria (NTM) include all mycobacteria species that do not belong to Mycobacterium tuberculosis (Mtb). They are a heterogeneous group of bacteria that are ubiquitously distributed in diverse environments such as water, sewage, soil, air, dust, food, animals and humans [1, 2]. To date, more than 150 species of NTM have so far been identified, but most of them are still poorly characterized and their clinical relevance is poorly understood.
In recent years, a number of species of NTM have been found to be associated with a wide range of human infections such as pulmonary, cutaneous and disseminated diseases [3, 4] They also pose a threat to people with various underlying medical conditions such as cystic fibrosis, bronchiectasis, prior tuberculosis and immune suppression [3, 4]. The emergence of the HIV/AIDS epidemic in the past few decades has resulted in the rise of NTM infections in the human population [4]. Together with Mycobacterium tuberculosis, the NTM species are common opportunistic infections in people with HIV/AIDS [5, 6].
Reliable and sensitive laboratory methods for identification of infection by NTM species are therefore normally required. The identification of NTM using traditional methods such as culture and biochemical analysis is not very sensitive and specific. Molecular methods such as polymerase chain reaction and sequencing of specific genes have recently offered the best approach to NTM speciation [7, 8]. The objective of this study was to identify NTM species stored at NMRL in Harare which were previously isolated during a national TB survey in Zimbabwe using 16S ribosomal RNA gene sequencing.
2. MATERIALS AND METHODS
2.1. Study Design and Origins of NTM Samples
The research design was an observational study in which NTM samples previously isolated from humans throughout Zimbabwe during a national TB survey were used. The samples were already stored at the National Microbiology Reference Laboratory. These samples were collected during the national TB survey that was carried out by the Ministry of Health and Child Care of Zimbabwe in 2014. During sample collection, all adult subjects gave their verbal informed consent for inclusion before they participated in the study. Sputum samples were collected from participating subjects after consenting. Two methods were used to screen for participants who were enrolled into the study, a questionnaire and chest X-Ray. From the questionnaire data, anyone with a cough of any duration was recruited into the study. With the chest X-ray, any abnormality consistent with TB was also considered as an entry point into the study. Two sputum samples, spot and morning, were collected from each of the participants and sent to the NMRL under cold chain conditions for processing. The two sputum samples from each participant were processed for TB culture and inoculated onto solid media, Lowensen Jensen (LJ) media (Becton Dickson Microbiology Systems, USA) and liquid media, Mycobacterial Growth Indicator Tube (MGIT) (Becton Dickson Microbiology Systems, USA). LJ culture tubes were incubated at 37°C while MGIT tubes were incubated in the BACTEC MGIT 960 machine and growth monitored. Once growth was observed on LJ and the BACTEC MGIT 960 Flagged positive, samples were stained with Ziehl Neelsen stain. If acid fast bacilli were present, the isolates were tested for presence of Mycobacterium tuberculosis (Mtb) bacteria using a rapid identification kit for Mtb, the MPT64 Ag kit (Standard Diagnostics, Korea) according to manufacturer’s recommendations. Positive results indicated presence of Mtb while negative results indicated the NTM. However to prove they were NTM, the isolates were further tested for growth in presence of paranitrobenzoic acid and their ability to grow at different temperatures, 25°C, 37°C and 45°C. Cultures were stored in liquid media at -80 oC until DNA extraction.
2.2. DNA Extraction from NTM Isolates
To prepare DNA extracts, 1ml of bacteria grown in liquid media was collected and centrifuged (with centrifuge with aerosol tight rotor) for 15 min at 10 000g. The supernatant was discarded and 100 µl sample lysis buffer (Hain Lifescience, Germany) added. The pellet was resuspended in the buffer and incubated at 95°C in a heat-block. After incubation for 5 mins, 100 ul of sample neutralization buffer (Hain Lifescience, Germany) was added and the mixture briefly vortexed. The extract was centrifuged for 5 mins at maximum speed in a centrifuge with aerosol tight rotor. The supernatant (DNA extract) was transferred into new tube and stored at -20°C until polymerase chain reaction of the 16S rRNA gene.
2.3. Polymerase Chain Reaction of the 16S rRNA Gene, Sequencing and Bioinformatics
The 5'-end of the 16S rRNA gene of genus Mycobacterium was amplified by polymerase chain reaction as previously described [9]. The Mycobacterium-specific forward primer, 5′-CCT GCA CTT CGG GAT AAG CCT G -3′, and reverse primer, 5′-CAA CGC GAC AAA CCA CCT ACG A -3′ were used for the polymerase chain reaction. The following cycling program was used for amplification: initial denaturation of 5 minutes at 95°C followed by 35 amplification cycles of 30 seconds at 95°C, 30 seconds at 55°C, and 45 seconds at 72°C, and ending with a final extension step of 7 minutes at 72°C. PCR products (10 ul) from the samples that amplified well and with a gel band of approximately 460 bp were sent for sequencing at Inqaba Biotechnical Industries, South Africa. Only the forward primer was used for the sequencing of the amplicons. DNA sequences were analyzed using Geneious Basic program (Biomatters, USA) and the Basic Local Alignment Search Tool (BLAST) programs. The different NTM species were identified using Geneious Basic program which compared our sequences with those in the Genbanks. Phylogenetic trees were also constructed using the Geneious Basic program.
3. RESULTS
In the Zimbabwe National TB prevalence survey of 2014, a total of 963 NTM samples were isolated from the participants and stored at the National Microbiology Reference Laboratory in Harare. In this study, we retrieved 99 of the 963 isolates for speciation using 16S ribosequencing molecular method.
Polymerase chain reaction of the 16S ribosomal RNA gene was successfully performed on the 99 NTM DNA samples (Fig. 1). An expected DNA band of approximately 460 bp was observed on agarose gel for most samples. Out of the 99 samples, 94 showed successful amplification of the ribosomal RNA gene. Of these 94 amplicons, 90 were sequenced. Bioinformatic analysis was performed on 81 of the NTM samples that showed perfect sequence results.

A representative gel of 16S ribosomal DNA amplicons from 9 NTM isolates. M is the molecular weight marker; lanes 1 to 8 show positive NTM rRNA gene amplicons, and lane 9 showed no amplification of the NTM rRNA gene. The expected PCR band for the 5¢-end of the 16S rRNA was approximately 460 bp.
Analysis of the DNA sequences of the 81 NTM isolates using bioinformatics revealed interesting findings. From the 81 sequence samples analyzed, 40 (49.4%) belonged to the Mycobacterium avium-intracellulare complex (MAC) species and the other 41 (50.6%) were non-MAC species (Table 1). The 40 samples (coded: B3, B5, B7, B8, B19, B21, B22, B25, B26, B27, B28, B29, B30, B32, B33, B36, B37, B39, B40, B41, B42, B43, B44, B45, B46, B48, B49, B51, B54, B56, B61, B63, B65, B67, B68, B70, B73, B76, B81, B88) had (except one) sequence similarity of 100% with at least one of the MAC species found in Genbank.
The MAC species that had sequence similarities with our samples were mainly M. avium, M. intracellulare, M. colombiense, M. yongonense, M. marseillense, M. vulneris and M. indicus from the Genbank. Further phylogenetic analysis of the 40 MAC samples showed minor sequence variations (Fig. 2). Some of the sequences were 100% similar, while others differed by few bases. Sequences B7, B19, B28, B29, B32, B33, B40, B41, B42, B46, B65 and B75 were identical and they had 100% sequence similarity with M. intracellulare and M. vulneris from the Genbank. Another group of sequences (B3, B8, B25, B21, B27, B39, B43, B54, B56, B88) had 100% similarity with M. avium, M. intracellulare, M. yongonense, M. colombiense, M. bouchedurhonense. Sequences B26, B37, B44, B51, B61, B68 were 100% similar with those of M. avium, M. intracellulare, M. yongonense, M. marseillense. Sequences of B5, B22, B36, B48, B76 were similar with those of M. avium, M. intracellulare, M. yongonense, M. colombiense and M. bouchedurhonense in the Genbank. Sequences of B63, B67, B70, B81 were similar with only those of M. avium and M. intracellulare. Sequences of B30, B45, B49 were closely related, but differed slightly and were similar with a number of MAC species in the Genbank.
Identification of species of NTM by 16S ribosequencing (n = 81).
| Mycobacterium Species | No. of strains identified (%) | Sample sequence code (and %identity) |
|---|---|---|
| M. avium-intracellulare complex (MAC) | 40 (49.4%) | B3 (100%), B5 (100%), B7 (100%), B8 (100%), B19 (100%), B21 (100%), B22 (100%), B25 (100%), B26 (100%), B27 (100%), B28 (100%), B29 (100%), B30 (100%), B32 (100%), B33 (100%), B36 (100%), B37 (100%), B39 (100%), B40 (100%), B41 (100%), B42 (100%), B43 (100%), B44 (100%), B45 (100%), B46 (100%), B48 (100%), B49 (99.7%), B51 (100%), B54 (100%), B56 (100%), B61 (100%), B63 (100%), B65 (100%), B67 (100%), B68 (100%), B70 (100%), B73 (100%), B76 (100%), B81 (100%), B88 (100%) |
| M. lentiflavum | 5 (6.2) | B2 (100%), B4 (100%), B18 (100%), B23 (100%), B58 (100%) |
| M. terrae complex | 4 (4.9) | B13 (100%), B24 (99.9%), B47 (100%), B50 (100%) |
| M. paraense | 4 (4.9) | B9 (100%), B12 (99.7%), B52 (99.9%), B72 (100%) |
| M. kansasii | 3 (3.7) | B11 (99.5%), B20 (99.5%), B55 (99.5%) |
| M. moriokaense | 3 (3.7) | B74 (99.2%), B80 (99.2%), B90 (99.1%) |
| M. asiaticum | 2 (2.5) | B16 (100%), B34 (99.3%) |
| M. novocastrense | 2 (2.5) | B31 (100%), B57 (100%) |
| M. brasiliensis | 2 (2.5) | B38 (99.2%), B75 (100%) |
| M. elephantis | 2 (2.5) | B79 (100%), B86 (100%) |
| M. paraffinicum | 1 (1.2) | B10 (99.9%) |
| M. bohemicum | 1 (1.2) | B14 (99.6) |
| M. manitobense | 1 (1.2) | B15 (99.0%) |
| M. intermedium | 1 (1.2) | B35 (100%) |
| Mtb complex | 1 (1.2) | B62 (100%) |
| M. parakoreense | 1 (1.2) | B66 (99.0%) |
| M. florentinum | 1 (1.2) | B71 (100%) |
| M. litorale | 1 (1.2) | B82 (100%) |
| M. fluoranthenivorans | 1 (1.2) | B83 (100%) * |
| M. sherrisii | 1 (1.2) | B84 (100%) |
| M. fortuitum | 1 (1.2) | B85 (100%) |
| M septicum | 1 (1.2) | B87 (100%) ** |
| Unidentified species | 2 (2.5) | B64 (98.8% M. montefiorense), B69 (98.6% M. phlei) |
| TOTAL | 81 (100) |
*was also 100% similar with M. neoaurum and M. cosmeticum, **was 100% similar with M. porcinum.
The other Mycobacterium species (n = 41) (50.6%) other than MAC that were identified in this study were M. lentiflavum (6.2%), M. terrae complex (4.9%), M. paraense (4.9%), M. kansasii (3.7%), M. moriokaense 3.7%), M. asiaticum (2.5%), M. novocastrense (2.5%), M. brasiliensis (2.5%), M. elephantis (2.5%), M. paraffinicum (1.2%), M. bohemicum (1.2%), M. manitobense (1.2%), M. intermedium (1.2%), M. tuberculosis complex (1.2%), M. parakoreense (1.2%), M. florentinum (1.2%), M. litorale (1.2%), M. fluoranthenivorans (1.2%), M. sherrisii (1.2%), M. fortuitum (1.2%) and M septicum (1.2%) (Table 1). Two isolates (B64 and B69) could not be identified, but were found to be closely related to M. montefiorense and M. phlei respectively (Table 1). Phylogenetic analysis of the 41 sequences showed that some of the isolates were closely related while others were distantly related (Fig. 3).

The 16S rRNA gene-based phylogenetic tree analysis of the 40 MAC species. The tree was rooted with Mtb sequence (Accession number KF796660).
4. DISCUSSION
NTM are emerging pathogens in the human population especially in resource-limited countries [10]. They are now known to cause a number of opportunistic infections in both immunosuppressed and immunocompetent individuals [11]. No previous studies have been done to identify NTM species prevalent in the human population in Zimbabwe. In this study, we set out to identify NTM species circulating in Zimbabwe. We therefore used NTM samples that were stored at the NMRL and were previously collected during the national TB survey in 2014 by the Ministry of Health and Child Care. The main objective of the survey had been to determine the prevalence of Mtb in Zimbabwe. However, during the survey, NTM bacteria were also isolated. The use of ribosomal genes of bacteria such as 16S, 5S and 23S in species identification has gained momentum in the past few decades [12, 13]. These genes have domains which are species-specific and hence can be used in speciation. 16S ribosequencing is the most commonly used method in identification of species of NTM from various sources [14, 15]. Other different methods which are also used in identifying NTM species include sequencing of rpoB, hsp65 and sod genes [7, 16].
This study highlighted the existence of several NTM species in the human population in Zimbabwe. Species belonging to the MAC group were the commonest in this study. The MAC group of NTM is made up of closely related Mycobacterium species, mainly M. avium and M. intracellulare, together with other few less-defined species such as M. colombiense, M. yongonense, M. marseillense, M. vulneris, M. indicus and M. paratuberculosis. These species are ubiquitous in environmental sources such as natural and municipal waters, soil, animals and droppings of infected birds [17]. Humans can get infected from these contaminated sources. It is now known that MAC species are responsible for the majority of infections caused by NTM in both developed and developing countries. Both immunocompetent and immunocompromized human populations are at risk of getting infected opportunistically by MAC species [10, 11, 18, 19]. People who have HIV/AIDS have a higher risk of getting infected [20]. Generally, infections due to Mycobacterium tuberculosis and MAC are the most commonly reported in HIV/AIDS patients [21, 22].
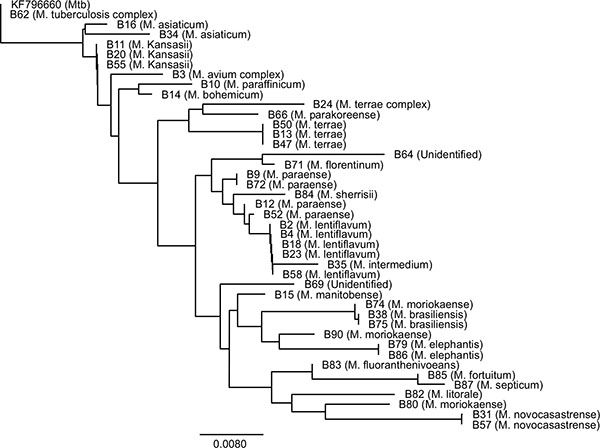
The 16S rRNA gene-based phylogenetic tree analysis of the 41 non-MAC species. The tree was rooted with Mtb sequence (Accession number KF796660). B3 was used as a representative of the MAC species.
Other than MAC, we also identified other Mycobacterium species that could be of human health importance. They were M. lentiflavum, M. terrae, M. paraense, M. kansasii, M. moriokaense, M. asiaticum, M. novocastrense, M. brasiliensis, M. elephantis, M. paraffinicum, M. bohemicum, M. manitobense, M. intermedium, M. tuberculosis complex, M. parakoreense, M. florentinum, M. litorale, M. fluoranthenivorans, M. sherrisii, M. fortuitum and M septicum (Table 1). In this study, there was no clinical data such as HIV status of the subjects where the NTM were isolated. The clinical relevance of the NTM species identified in Zimbabwe could not therefore be ascertained. However, the species we identified were found to be associated with specific infections in other studies. M. lentiflavum which is considered to be an emerging human pathogen has been found in children with cervical lymphadenitis and in immuno compromised patients [23, 24]. M. lentiflavum can also cause chronic lung disease in immuno competent people [25]. The M. terrae complex (M. triviale, M. nonchromogenicum, and M. terrae) can cause some minor diseases such chronic bronchopulmonary disease, lymphadenitis, skin and soft tissue disease, skeletal (bone, joint, and tendon) disease, and disseminated and catheter-related infections [26]. M. nonchromogenicum has been associated with arthritis, tenosynovitis, meningitis and pulmonary infection [27]. M. paraense was previously isolated from pulmonary samples from Brazil, but its clinical importance is still poorly known [28]. M. kansasii is a photochromogenic bacterium and is known to be the second commonest (after MAC) cause of NTM disease in HIV/AIDS patients [22, 29]. M. moriokaense was found to be associated with chronic pneumonia, cutaneous and wound infections [30, 31]. This rare NTM species was recently isolated from water supplies [32]. M. asiaticum is regarded as a cause of pulmonary disease, childhood lymphadenitis, olecranon bursitis and wound infections [33]. M. moriokaense species was shown to be associated with chronic pneumonia [34]. Clinical importance of Mycobacterium brasiliensis is still scarce. M. elephantis was originally detected from a sick elephant that that died from chronic respiratory disease and is associated with respiratory diseases in humans [35]. M. paraffinicum has been associated with symptomatic pulmonary infections [36]. M. bohemicum has been associated with cervical lymphadenitis in children [37]. M. manitobense is a rapidly growing mycobacterium that is potentially pathogenic to humans [38]. M. intermedium has been found in cutaneous and pulmonary infections [39, 40]. One sample (B62) aligned with sequences of the Mtb complex (M. tuberculosis, M. bovis, M. aficanum, M. canetti and M. microti). The sequencing of 16S ribosomal RNA gene cannot discriminate species of the Mtb complex and hence more molecular methods would be required to identify the species [16, 41]. M. parakoreense has been associated with pulmonary infections [42]. M. florentinum has been detected in people with various pulmonary disorders and cervical lymphadenitis [43, 44]. M. litorale was recently isolated from soil and its medical importance is not yet studied [45]. M. fluoranthenivorans is a fluorenthene and aflotoxin-degrading mycobacterium and has no known clinical relevance [46]. M. neoaurum is known to be associated with bacteraemia and other pulmonary infections [47, 48]. M. cosmeticum is an opportunistic pathogen in humans that is associated with cutaneous granulomatous lesions and gastrointestinal tract problems [49, 50]. M. sherrisii has been associated with pulmonary and disseminated disease in HIV/AIDS patients [51, 52]. M. fortuitum was sometimes associated with catheter infections [53]. M. septicum was previously found in sepsis and bacteraemia [54, 55]. In this study, two isolates could not be identified. Further analysis showed that they were closely related to M. montefiorense and M. phlei respectively. M. montefiorense causes skin lesions in moray eels [56]. Its clinical relevance to humans is still to be established. M. phlei is widely distributed in the environment and can cause opportunistic infections [57].
The sources of human infections by NTM in Zimbabwe were not investigated in this study. Some epidemiologic studies elsewhere have suggested that water (natural and municipal water) was one of the main sources of human exposure to NTM [58]. Water has also been found to be the main source of M. avium [59, 60]. Other sources of human infections include soil, sewage and wastewater, aerosols, plants, food, protozoa, animals, birds and humans [1, 2, 4, 61, 62]. It is therefore the overlap of human and mycobacterial ecology that will continue to make NTM infections in the human population unavoidable [4]. A number of further studies will need to be done to establish the scope of the NTM problem in Zimbabwe.
CONCLUSION
The study highlighted the importance of identifying species of NTM in Zimbabwe. The MAC species were found to be the most common NTM in humans during the national TB survey in Zimbabwe. Further studies are necessary to ascertain the true prevalence and clinical relevance of NTM species in the Zimbabwe especially in people with HIV/AIDS. Further studies are also necessary to establish the sources of human infections by NTM.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The authors wish to thank colleagues at the Department of the Medical Microbiology of the University of Zimbabwe and the National Microbiology Reference Laboratory for assistance during the study. The research study was funded by the University of Zimbabwe’s Research Board Project Grant Code 91065. The national TB survey was conducted by the Ministry of Health and Child Care of Zimbabwe and the National Microbiology Reference Laboratory.

