All published articles of this journal are available on ScienceDirect.

In Silico Design of a Chimeric Protein Containing Antigenic Fragments of Helicobacter pylori; A Bioinformatic Approach
Authors Info & Affiliations
Abstract
Helicobacter pylori is a global health problem which has encouraged scientists to find new ways to diagnose, immunize and eradicate the H. pylori infection. In silico studies are a promising approach to design new chimeric antigen having the immunogenic potential of several antigens. In order to obtain such benefit in H. pylori vaccine study, a chimeric gene containing four fragments of FliD sequence (1-600 bp), UreB (327-334 bp),VacA (744-805 bp) and CagL(51-100 bp) which have a high density of B- and T-cell epitopes was designed. The secondary and tertiary structures of the chimeric protein and other properties such as stability, solubility and antigenicity were analyzed. The in silico results showed that after optimizing for the purpose of expression in Escherichia coli BL21, the solubility and antigenicity of the construct fragments were highly retained. Most regions of the chimeric protein were found to have a high antigenic propensity and surface accessibility. These results would be useful in animal model application and accounted for the development of an epitope-based vaccine against the H. pylori.
INTRODUCTION
H. pylori is a gram negative microaerophilic bacterium which inhabits the mucus layer overlying gastric epithelial cell [1-3]. The resulting infection may cause mild to severe complications including gastritis, peptic ulcer, gastric adenocarcinoma and gastric mucosa-associated lymphoid tissue lymphoma (MALT) [4, 5]. This microorganism infects half of the world's population and is considered as a type I carcinogen by the World Health Organization [6]. Although antibiotic therapy is effective in the majority of patients with gastrointestinal symptoms, current therapies such as the use of proton pump inhibitors combined with antibiotics, has several disadvantages including low patient compliance, high cost and increased antibiotic resistance. Consequently, the treatment by antibiotics aren’t ideal [7]. Therefore, an alternative approach is needed to overcome the problems of the conventional treatment of H. pylori infection. Synthetic vaccines can offer long-term solutions to numerous important infectious diseases. So an effective vaccine would require to prohibit H. pylori-induced gastric disease. For achieving the mentioned therapeutic goals, the subunit vaccine is a promising way to increase the safety. But the development of such vaccines depends on the identification of antigens able to confer protection against H. pylori. Numerous protein antigens of H. pylori were evaluated including Urease [8-13] , heat-shock proteins [14, 15], VacA cytotoxin [16], thiolperoxidase (Tpx) [17], OipA [18], HpaA [19], NapA [20], and CagA [16, 21]. Previous studies illustrated that fusion proteins, a class of novel engineered biomolecules, have multi-functional properties derived from each of their component moieties [22, 23], therefore, combining various antigens participating in different aspects of colonization and pathogenesis having enough potential to elicit a board of cellular or humoral immune response that may be more effective than a single recombinant antigen inducing insufficient immunity with limited protective effect [24-27]. There is an increasing interest to determine the potential of therapeutic or protective effect of such fusion proteins. In this regard, some chimeric proteins obtained from different components of H.pylori have been proposed including HpaA, UreB and CagA as a fusion protein stimulating CD4+ T cell response [28]. Furthermore, in another study three protective antigens including UreB, VacA and CagA were used to construct a new fusion protein expressed in live attenuated Salmonella showing increased the levels of IgG2a, mucosal IgA and induced Th1 phenotype response [29]. However, how some antigenic fragment can be combined as they show their highest immunogenicity and protectivity effect would be a problem. Bioinformatics is an interdisciplinary area combining statistics, computer science, mathematics, and engineering to evaluate and interpret of data in order to improve methods and software tools for understanding biological data. Fortunately, by using such interdisciplinary approach, we would have the chance to understand the function of a designed chimeric protein in vivo. Then, bioinformatic approach would help us effectively design a chimeric protein with more immunogenicity and protectivity.
In this study, we postulated that combination of FliD with antigenic fragments of well-known H.pylori proteins including UreB, VacA and CagL may enhance the cumulative antigenicity of antigens.
FliD protein (685aa) is an essential element in assembling of the functional flagella and a FliD mutant strain is completely non-motile. Flagellin plays a central role in bacterial motility and is necessary for colonization and persistence of H. pylori infection [30]. Interestingly, It has been shown that FliD protein reacts approximately with 97 percent of sera obtained from patients infected with H.pylori [31].
Urease B has been widely investigated as a potential antigen for the development of prophylactic and therapeutic vaccines against H. pylori infection [32, 33]. UreB(327-334) is considered as a good B cell epitope and has been found to be protective in mice [34, 35].
Another well-characterized H. pylori protein is the vacuolating cytotoxin (VacA). It induces large cytoplasmic vacuoles and apoptosis which are partly responsible for H. pylori induced epithelial cell damage [36, 37]. It suggests that 744-805aa of VacA includes MHC Class-II binding regions which efficiently stimulates CD4+ T-cells responses [29].
CagL protein is a versatile type IV secretion system (T4SS) surface protein equipped with at least two motifs to help binding to integrins causing the deviant signaling within host cells and facilitating translocation of CagA into host cells [38]. The critical role of CagL in attaching to the epithelial cells and starting pathogencity of H. pylori [39] may make it a good candidate for further studies.
To test our postulate, we designed a new structural model containing most dominant epitopes of FliD, UreB, VacA and CagLthat may provide a proper and safe candidate for prophylaxis against H.pylori infection. In this regard, FliD, UreB, VacA and CagL, were bound together by three hydrophobic linkers. Then, chimeric protein structure was analyzed through a bioinformatic approache.
MATERIALS AND METHODS
Sequence Retrieval and Arrangement
Sequences of antigens were obtained from GenBank. In order to identify the general and conserved antigenic fragments among all sequences, multiple sequence alignments were performed using ClustalW software (www.ebi.ac.uk/Tools/msa/clustalw2/). Afterward, the multi parameter characterizations and basic physico-chemical parameters of the chimeric protein such as molecular weight, extinction coefficient, half-life, theoretical isoelectric point (pl), etc., were computed using stand-alone software, such as Gene Designer v.1.1 (www.dnatwopointo.com) and Prot-Param (www.dnatwopointo.com); (expasy.org/tools/protparam.html).
Codon Optimization
In order to identify a fragment common among all sequences, multiple sequence alignments were performed using ClustalW software (http://www.ebi.ac.uk/Tools/clustalw2). Stand-alone Leto gene optimization software (www.entelechon.com), OPTIMIZER server [40], Kazusa codon usage database (www.kazusa.or.jp/codon) and the reverse translation online tool (www.bioinformatics.org/sms2/rev_trans.html) were used for the in silico gene analysis and optimization of the chimeric gene. Another online software used to optimize our sequences was JavaScript programs for analyzing and formatting protein and DNA sequences (www.ualberta.ca/~stothard/software.html) [41]. The chimeric gene was designed for cloning and expression in E. coli BL21. VaxiJen server was used to predict the immunogenicity of the whole antigen and its subunit vaccine [42]. The multimeric gene was synthesized by Biomatik (Ontario, Canada). Some parameters such as codon adaptation index (CAI), frequency of optimal codon (FOP) and GC content are critical to the efficiency of gene expression. In addition to the removing of mRNA instability motifs, adverse cis-acting components in the chimeric construct such as internal poly A and poly T sites, cryptic splice sites, repeat sequences and destabilizing that may decrease the expression level were also eliminated. The essential restriction enzyme sites ( HindIII, EcoRI) were presented at the ends of the sequence and other restriction sites were removed from the synthetic construct for the subsequent purpose of cloning. TAA, an effective stop codon, was used to increase the efficiency of translational termination in the prokaryotic host.
Prediction of mRNA Secondary Structure
For the high level expression in E.coli BL21, the chimeric gene optimized and EcoRI and HindIII restriction sites were added to the 5 and 3-ends of the sequence for the purpose of cloning. The structure of folding RNA messenger of the chimeric gene was analyzed before and after gene optimization and the energetic stability of the predicted structures compared together by the Mfold Web-based software (mfold.rna.albany.edu) [43], the RNAfold online server (rna.tbi.univie.ac.at) and GeneBee (www.genebee.msu.su/services/ rna2_reduced.html). Results were confirmed by CentroidFold Web Server, indicated the mRNA was stable enough for effectual translation in the prokaryotic host [44].
Prediction of Secondary and Tertiary Structures of the Chimeric Protein
The prediction of secondary structure of the chimeric protein was performed using GOR-IV (gor.bb.iastate.edu/) [45], PORTER (distill.ucd.ie/porter/) [46], SOPMA npsapbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npssopma.html [47] online servers. In order to analyze the sequence and predict the protein structure and function, the percentage of random coils, alpha-helices and beta sheets, the solvent accessibility were evaluated by the PredictProtein server [48].
The stability of the 3D structure of the chimeric protein was evaluated by Swiss-PdbViewer (swissmodel.expasy.org/workspace/) [49] and online softwares, such as I-TASSER (zhanglab.ccmb.med.umich.edu/I-TASSER/) [50], the Robetta server (robetta.bakerlab.org) and the LOOPP server (cbsuapps.tc.cornell.edu/loopp.aspx) [51] were used to predict the 3D structure of each selected segment and the complete designed construct. The validity of the predicted 3D structure was evaluated by the ProSA Web server [52] and was calculated by the Ramachandran plot drawn by the RAMPAGE (mordred.bioc.cam.ac.uk) and also by a comprehensive way of checking the geometry of the model, PROCHEK server (swissmodel.expasy.org/workspace). For picturing of three dimensional models of the chimeric protein, Rasmol tool and Accelrys Discovery Studio1.7 were used. The GROMOS96 implemented in SWISS MODEL software helps to reduce the bond str etc. h energy of the modeled protein [53].
Protein Solubility Prediction
The solubility of hydrophobic and polarity properties of different residues were evaluated by DSSP and other online programs such as VADAR (redpoll.-pharmacy.ualberta.ca/vadar/) [54] , the PROSO II server (mips.helmholtz-muenchen.de/prosoII). Furthermore, the prediction of the mean residue accessible surface area (ASA) in the chimeric protein was performed by the NetSurfP server (www.cbs.dtu.dk/services/NetSurfP/) [55].
Alergenic Sites Prediction
Prediction of the the IgE epitopes and allergenic potential of the chimeric protein were assessed by submitting the sequences to the Algpred server (www.imtech.res.in/ raghava/algpred/) [56] and APPEL servers (jing.cz3.nus.edu.sg/cgi-bin/APPEL). Further analysis of allergenicity was done by SDAP database (Structural Database of Allergenic Proteins) that can be used to predict allergenicity of novel proteins and cross-reactivity between allergens [57].
Prediction of Immunogenic Epitopes
B-cell Epitopes
B-cell epitopes are the sites of molecules that are recognized by antibodies of the immune system. The knowledge of B-cell epitopes may be used in the design of a good subunit vaccine [58]. Continuous (linear) B cell epitopes were obtained from BcePredsoftware (www.imtech.res.in/raghava/ bcepred) [59] and ABCpred servers (www.imtech.res.in/raghava/ abcpred/) [60]. Moreover, discontinuous (conformational) B cell epitopes were predicted using the DiscoTope 2.0 server (www.cbs.dtu.dk/services/DiscoTope/) [61]. Fortunately, a web server CBTOPE (www.imtech.res.in/raghava) has been developed for predicting of conformational B-cell epitopes in an antigen whose tertiary structures are not available and we also used this online software to predict the epitopes [62]. The BcePred program was used to determine the linear B cell epitope based on single characters such as antigenic propensity, polarity, flexibility, exposed surface, accessibility and hydrophilicity (Table 1). Totally, epitopes having BcePreds and VaxiJen cutoff values >0.8 and >0.4 are selected, respectively. VaxiJen was used to predict the immunogenicity of the whole antigen (www.jenner.ac.uk/VaxiJen) [63, 64]. Moreover, the location of conformational epitopes on protein surface was defined by Episearch software [64].
Thermodynamic features of mRNA optimized sequence.
| Structure | Energy | Position |
|---|---|---|
| Stack | -2.10 | External closing pair is U2-A2335 |
| Stack | -3.30 | External closing pair is G3-C2334 |
| Stack | -3.40 | External closing pair is G4-C2333 |
| Helix | -8.80 | 4 base pairs. |
T-cell Epitopes
In order to identify common epitopes that bind to both MHC class molecules as well as to count the total number of interacting MHC alleles, NetCTL and NetCTLpan were used. Such Softwares are predictors of T cell epitopes along a protein sequence. These tools at IEDB (www.cbs.dtu.dk/services/NetCTL predict the half maximal (50%) inhibitory concentration (IC50) values for peptides binding to specific MHC molecules. In addition, MHCPred (www.jenner.ac.uk/MHCPred) and SYFPEITHI servers (www.syfpeithi.de/bin/ MHCServer.dll/EpitopePrediction.htm) to predict epitope candidates based on the processing of peptides in the cell were also used [65, 66].
RESULTS
Design and Construction of Chimeric Gene
No redundant gene and related protein sequences of H.pylori virulence factors (FliD, UreB , VacA and CagL) were obtained from GenBank and analyzed with the ClustalW software (www.ebi.ac.uk/Tools/msa/clustalw2/) [67]. The nominated sequences for designing of chimeric construct were FliD (1-600), UreB (327-334), VacA (744-805) and CagL (51-100). These fragments were fused together using a repeat of hydrophobic amino acid linker. Four repeated GGGGS sequences were introduced between different domains for more flexibility and efficient separation. Schematic diagram of protein domain structures, their junctions and linker sites are shown in Fig. (1). A synthetic sequence encoding the chimeric protein was reverse translated into the nucleotide sequence. The Percentage of codons having a high-usage frequency distribution (91-100%) which was improved to 64 % in the optimized gene sequence (Fig. 2a). The codon usage bias in E.coli BL21 was increased by upgrading the codon adaption index (CAI) to 0.84 (Fig. 2b). The overall GC (guanine plus cytosine) content was enhanced to 50.33 in our antigenic sequence (Fig. 2c), which is a measurement of transcriptional and translational efficiency influenced as a compositional bias on the codon usage of a gene of interest. The Eco RI and HindIII restriction sites for cloning in prokaryotic vectors were successfully introduced at the N and C terminal of sequence, respectively.

Schematic representation of antigenic construct consists of FliD, UreB, VacA, CagL, attached together by linkers for eventual expression in prokaryotic cells.
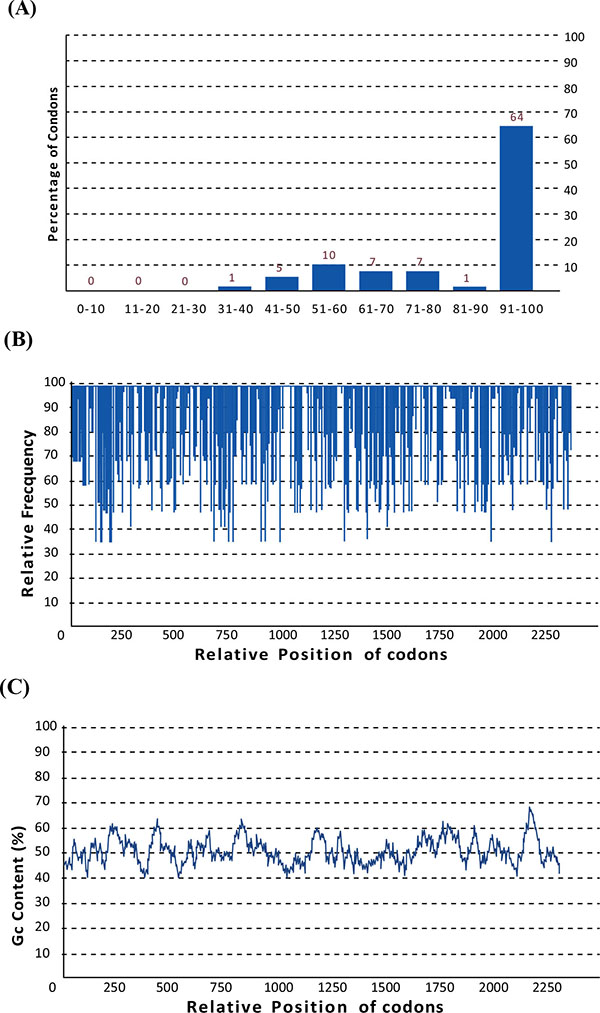
Codon usage bias adjustment of optimized gene for expression in E.coli (A); The percentage distribution of codons in computed codon quality groups. The value of 100 is set for the codon with the highest usage frequency for a given amino acid in the desired expression organism. Codons with values lower than 30 are likely to hamper the expression efficiency (B); The distribution of codon usage frequency along the gene sequence. A CAI of 1.0 is measured to be perfect in the desired expression organism, and a CAI of >0.8 is considered as good, in terms of high gene expression levels (C); GC content analysis of optimized chimeric gene; the ideal percentage range of GC content is between 30% to 70%. Any peaks outside of this range will adversely affect transcriptional and translational efficiency.
Prediction of mRNA Secondary Structure
The evaluation of minimum free energy for 37 structures of chimeric mRNA, unoptimized and optimized sequences carried out by the ‘Mfold’server. The results showed that ΔG of the best predicted structure for the optimized construct was ΔG = -745.80 kcal/mol. The first nucleotides at 5' did not have a long stable hairpin or pseudoknot. Therefore, the binding of ribosomes to the translation initiation site and the following translation process can be readily accomplished in the target host. These outcomes were in the agreement with data obtained from the ‘RNAfold’web server (Fig. 3). Thermodynamic features of the optimized sequence are shown in Table 1.
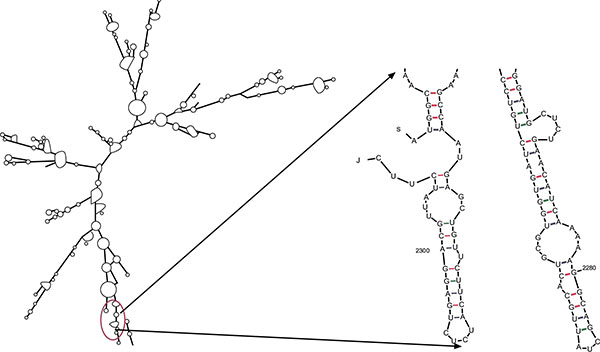
Analysis of mRNA stability and start codon in the structure.
The Physico-chemical Parameters
The basic physico-chemical parameters of the chimeric protein such as molecular weight was 84717.8 Da. Extinction coefficient of optimized chimeric protein at 280 nm was 30955M-1 cm-1. The bio computed half-life was greater than 10 hr. Acidity of the protein was shown by the pI value, pI: 5.01. Aliphatic index of synthetic chimeric protein was 85.34. Expasy's ProtParam classified the optimized the chimeric protein as stable (Instability index: 28.09).
Secondary Structure Prediction
The secondary structure of the chimeric protein was predicted by several online programs, and the best result was achieved by GOR-IV as shown in Fig. (4). Results indicated total residues of 785 which were made up 140 strands (17.83%), 299 helices (38.09%) and 346 random coils (44.08%). No predicted signal peptide was identified in the initial region of the protein sequence.
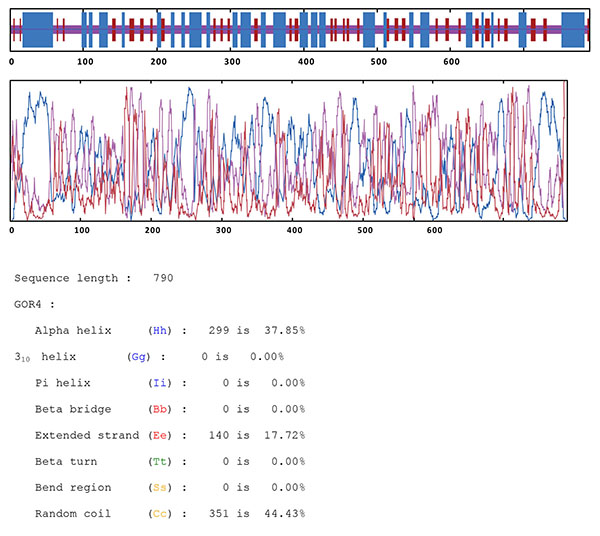
Analysis of chimeric protein secondary structure.
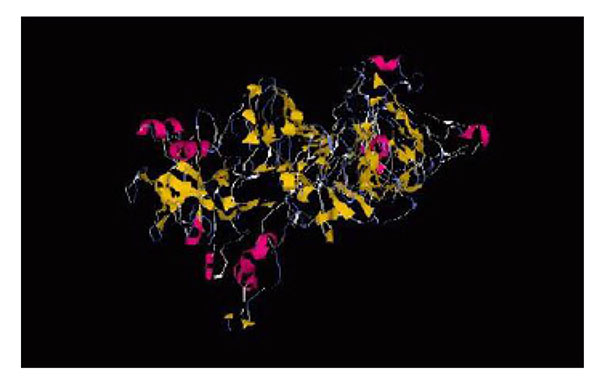
The tertiary structure of the chimeric protein, FliD-UreB-VacA-CagL.
Prediction of Tertiary Structure
Three dimensional structures of chimeric protein were predicted by I-TASSER server. The best model was selected based on the confidence score (C-score = -0.13) for estimating the quality of predicted model. C-score is typically in the range of (-5 to 2). Template modeling (TM-score = 0.70±0.12) for measuring the structural similarity between two structures. The results of tertiary structure prediction showed the formation of four separate domains of the chimeric protein (Fig. 5). The ProSA program was employed in the refinement and validation of the chimeric protein structures. The ProSA Web designated Z-score for the best predicted 3D model. The amount -0.73 was within the range of scores typically found for native proteins of the similar size.
Evaluation of Model Stability
The predicted model was subjected to energy minimization by application of the Swiss-PdbViewer. The amount -25414.018 kcal/mol indicated that the synthetic chimeric protein would have an acceptable stability compared the original structure of each moiety. The structure validity of the chimeric protein was assessed for its reliability and structural quality by the computation of a Ramachandran plot. Results of RAMPAGE showed that 65.6 % resides clustered in the favored, 24.5 % in the allowed and only 9.9% in the outlier (Fig. 6). The PROCHECK server verified the RAMPAGE results.

Evaluation of model quality based on the Ramachandram plot. Percentage of the residue was 65.6 % in the favored region (core beta), 24.5 % allowed (core-alpha) and 9.9% outlier (core left-handed alpha). It was designated that the backbone dihedral angles, φ and ψ, were reasonably accurate.
Protein Solubility Prediction
The solubility of the chimeric protein was interpreted using the PROSO II (mips.helmholtzmuenchen.de/prosoII) based on a classifier exploiting subtle differences between soluble proteins from TargetDB and the PDB and notoriously insoluble proteins from TargetDB. Our chimeric protein classified as soluble protein at threshold 0.6 with 71% accuracy. Residues that form the hydrophobic and polarity of the chimeric protein are used to characterize the solvent accessibility distributions.
Allergenic Sites Prediction
AlgPred tool was used to plot IgE epitopes of the sequence. There was no allergenicity found by MAST results. Based on various prediction approaches in the AlgPred tool and SDAP allergen library, this protein would not have any allergenicity and also not show significant similarities between any of the regions. As a final point, based on obtaining data from APPEL prediction tool working by comparing with sequence-derived structural and physicochemical properties of whole proteins, it was not found any allergenic protein.
Prediction of Immunogenic Epitopes
B-cell Epitopes
Using of BcePred Prediction Server, we could recognized linear B-cell epitopes based on physico-chemical properties of the chimeric protein (Table 2). ABCpred software predicted linear epitopes based on the artificial neural network method (Table 3). The CBTOPE server determining amino acid propensity scores and surface accessibility was used to predict discontinuous B cell epitopes (Table 4). VaxiJen predicted protective antigens and calculated the whole antigenicity of the chimeric protein with 0.7304 score, which was higher than 0.4 threshold.
Epitope predicted chimeric protein by physico and chemical properties based on Bcepred.
| Prediction Parameters | Epitopes Segments |
|---|---|
| Hydrophilicity | DKLKDADEKA, DKKMEQNVEKQK ,SRKSNVTGDA ,KFSSRDD ,TQNKDYA ,TDATNGEV ,MKTGGNDPYQ ,VNTKNTGEDNRI, GVDGSGKSEVS ,KGADGNM ,EQALENDPNFKD ,NDRRGGN ,KGSKAKE ,QTTTQESDL ,TKEGNTSEENTDAI ,QSYEASQNT ,QKASDSE ,EQTTEPNKPA ,SVNRDNQA ,PKLDEDTRYDADTK ,SVHTDNGVES ,NSNPKATQD ,YGSDSKDMGGRE ,HQEGGGGGSGGGGSDVQ ,SITSSDSQAMG ,QTADKNKKEFGGGGGSYN ,QGASYDN ,SNTNLQDQ ,NNNNRMD ,VRKNNTDDIKA ,GNQSGGGGSTTSPSANDKIGKED |
| Flexibility | DKLKDAD, ,STYISRKSNV, LGAKFSSRD, IVMKTGG, LMVNTKNTGED ,SLGVDGSGKSE ,ESASIKQ ,LIINDRRGGNIEIKGSKA, FLQTTTQESDLLKSSRT, KALTKEGNTSEEN, NAFKNAE ,LVINSKT ,LFMSKNLQKASD ,VSITRPT ,LEQTTEP ,AIISVNR ,GDIRTIRSSL ,DFFYGSDSKDMG ,EIHQEGGGGGSGGGG ,FSITSSDS ,TWQTADKNKKEFGGGG ,DNISASN, CVVRKNNT ,MAIGNQSGGGGSTTS |
| Accessibility | NYDVIDKLKDADEKA ,APLDKKMEQNVEKQKALVE ,KTLSDYSTYISRKSNVTG ,AKFSSRDD ,TLKFYTQNKDYAVN ,TDATNGE ,KTGGNDPYQLMVNTKNTGEDNRIYF ,QSTLTNKNALS ,DGSGKSE ,ESASIKQKNTAIQKAMEQALENDPNFKDL ,INDRRGGNIEIKGSKAKELG ,LQTTTQESDLLKSSRTIKEGKLEG ,GQKLDLKALTKEGNTSEENTDAI ,QAINAKEGLNAFKNAEGKL ,QSYEASQNT ,MSKNLQKASDSEFTYN ,SITRPTNEIND ,TLEQTTEPNKPAI ,SVNRDNQAI ,YNELIPKLDEDTRYDADTKI ,DEAKLSSALNSNPKATQDF ,YGSDSKDMGGREIHQEG ,ADSRIRPQTI ,TRTWQTADKNKKEFGG ,QGASYDN ,SNTNLQDQFKERLALYNNNNRMDI, VVRKNNTDDIKA ,TSPSANDKIGKEDA , KANFEAN |
| Turns | LENDPNFKD, SSLNNVFS ,SVHTDNGV ,ALNSNPKA, SASNTNLQ, ALYNNNNRMD, RKNNTDDI |
| Exposed Surface | DKLKDADEKA, PLDKKMEQNVEKQKA ,KFYTQNKDY, SIKQKNT ,ENDPNFKD, KGSKAKE ,KSSRTIKEGK, MSKNLQKAS ,QTTEPNKP, KLDEDTRYDADTK, QTADKNKKEFG ,QDQFKERL ,VRKNNTDD ,NDKIGKED |
| Polarity | DVIDKLKDADEKAL ,PLDKKMEQNVEKQKALVE, KFSSRDD, RRGGNIE ,KGSKAKE ,KSSRTIKEGKLEG, KEGNTSEE ,FKNAEGKL , IPKLDEDTRYDADTK, KDMGGREIHQEGG ,AEDTLHDM ,TADKNKKEFGG ,QDQFKERLALY,RKNNTDD ,NDKIGKEDA |
| Antigenic Propensity | SLSSLGL ,KVLNYDVIDKL ,LVEIKTLL ,KVDVQNL ,IYFGSHL, HEVPIMLEL, LEGVISL, VISGVNI, NVFSYSVHT, SYNLVGVQ , RMDICVVRKN |
The predicted linear B- cell epitopes by ABCpredserever.
| Rank | Sequence | Start position | Score |
|---|---|---|---|
| 1 | VGEVITRTWQTADKNK | 649 | 0.94 |
| 2 | GGSTTSPSANDKIGKE | 738 | 0.93 |
| 3 | QAIIDSLTEFVKAYNE | 482 | 0.91 |
| 3 | GFLQTTTQESDLLKSS | 309 | 0.91 |
| 4 | CGMAIGNQSGGGGSTT | 727 | 0.90 |
| 4 | TEFVKAYNELIPKLDE | 489 | 0.90 |
| 5 | TGGNDPYQLMVNTKNT | 172 | 0.88 |
| 6 | MSLDEAKLSSALNSNP | 561 | 0.87 |
| 6 | NTAIQKAMEQALENDP | 251 | 0.87 |
| 7 | TYNGVSITRPTNEIND | 439 | 0.86 |
| 8 | VKTLSDYSTYISRKSN | 64 | 0.85 |
| 8 | EGGGGGSGGGGSDVQF | 599 | 0.85 |
| 8 | VQAINAKEGLNAFKNA | 363 | 0.85 |
| 8 | QALENDPNFKDLIANR | 260 | 0.85 |
| 8 | NGEVMGIVMKTGGNDP | 162 | 0.85 |
| 9 | ADSRIRPQTIAAEDTL | 615 | 0.84 |
| 10 | PESASIKQKNTAIQKA | 242 | 0.83 |
T-cell Epitopes
NetCTL software was used to predict the T cell epitopes, 17 MHC ligands were identified at 0.75 threshold. In addition to the check of each T cell epitope by the SYFPEITHI server to identify T cell MHC class I and MHC class II , the MHC Pred software was also used to identify the common T cell epitopes (Table 5).
Conformational B-cell epitopes prediction based on CBTOPE server, which was higher than 3 threshold.
| Position | Epitope | Position | Epitope | Position | Epitope |
|---|---|---|---|---|---|
| 21-22 | ID | 294-8 | RGGNI | 555-60 | LDDKGV |
| 32-34 | LIA | 311 | L | 564 | D |
| 62 | G | 332 | L | 572-3 | LN |
| 66-70 | TLSDY | 338-9 | LN | 576-83 | PKATQDFF |
| 74 | I | 343 | L | 588-90 | SKD |
| 95 | I | 352-6 | GNTSE | 634-43 | GIFSITSSDS |
| 103-5 | QNL | 359-62 | TDAI | 647-56 | GRVGEVITRT |
| 109-13 | DINEL | 365-8 | AINA | 659-74 | TADKNKKEFGGGGGSY |
| 134-8 | YTQNK | 369 | E | 686-94 | NISASNTNL |
| 143-7 | NIKAG | 400-2 | GKA | 698-9 | FK |
| 152-6 | DVAQS | 405 | K | 702 | L |
| 158 | T | 409 | L | 704 | L |
| 162-4 | NGE | 414 | M | 718-31 | KNNTDDIKACGMAI |
| 170-1 | MK | 416 | S | 734 | Q |
| 176-8 | DPY | 435-9 | DSEFT | 736 | G |
| 180-99 | LMVNTKNTGEDNRIYFGSHL | 446 | T | 740-50 | STTSPSANDKI |
| 204 | T | 456 | I | 762 | A |
| 209 | L | 472-8 | PAIISVN | 768-71 | DLSL |
| 222 | S | 483 | A | ||
| 226-7 | KG | 487 | S | ||
| 258-60 | MEQ | 499 | I | ||
| 266 | P | 501-3 | KLD | ||
| 269 | K | 521-31 | VGDIRTIRSSL | ||
| 282 | L | 536-9 | SYSV | ||
| 288-90 | LII | 541-4 | TDNG | ||
| 292 | D | 549-51 | NKY |
The predicted T- cell epitopes by MHC Predserever.
| Amino acid groups | Predicted -logIC50 (M) | Predicted IC50 Value (nM) | Confidence of prediction (Max = 1) |
|---|---|---|---|
| SLMKYGLSL | 7.739 | 18.24 | 1.00 |
| IMLELPESA | 7.483 | 32.89 | 1.00 |
| IIDSLTEFV | 7.454 | 35.16 | 1.00 |
| SLLKANFEA | 7.123 | 75.34 | 1.00 |
| FVKAYNELI | 7.061 | 86.90 | 1.00 |
| ALKGPVKTL | 7.054 | 88.31 | 1.00 |
| SLKDLGLSA | 7.035 | 92.26 | 1.00 |
| DADTKIAGI | 6.899 | 126.18 | 1.00 |
| KLKDADEKA | 6.887 | 129.72 | 1.00 |
| ALRGDLSLL | 6.78 | 165.96 | 1.00 |
| KALVEIKTL | 6.682 | 207.97 | 1.00 |
| VISGVNITL | 6.643 | 227.51 | 1.00 |
| AIQKAMEQA | 6.635 | 231.74 | 1.00 |
| IALRGDLSL | 6.629 | 234.96 | 1.00 |
| MAIGSLSSL | 6.558 | 276.69 | 1.00 |
DISCUSSION
H.pylori is a microorganism that has been classified as class I human carcinogen and has been linked to diseases such as gastritis and peptic ulcers. It has a high incidence of infection, especially in developing countries and the antibiotic therapy is often not successful [6]. Taking these issues into account, the recombinant subunit vaccine may have a potential preventive strategy in H.pylori infection which would defiantly help to reduce the risk of such infection. In the current study, different bioinformatic approaches were employed for predicting B and T cell epitopes by using only those peptides that possess high immunogenicity instead of the whole proteins. Based on our finding, a chimeric protein including immunodominant epitopes from different antigenic proteins such as FliD (1-600), UreB(327-334), VacA (744-805) and CagL (51-100) would likely induce strong comprehensive protective immunity. These amino acid sequences were checked and found that these epitopes highly conserved among different H. pylori strains. Suitable linker to attach the multi-domain proteins is important for desired component protein and accurate function [68]. These different domains are fused together using a repeat of GGGGS (Gly-Gly-Gly-Gly-Ser) linker. Two GGGGS linkers are placed between FliD and UreB, entailing two GGGGS moieties between UreB and VacA, CagL. Flexible GS spacers could provide structural flexibility improving protein stability and increasing biological activity [23]. Additionally, it has been reported that suitable linker peptides might be more effective than independent domains to improve the biological activity or increase the protein stability [68-70]. The physico-chemical parameters of chimeric protein were analyzed. The theoretical isoelectric point (pI) values of the fusion protein, as calculated by the Prot-Param were detected to be at 5.01. Thus, the protein is considered to be stable and compact at pI. Molecular weight was 84717.8Da which provides information on the full-length of protein expression, expression of modified, cleavage products, splice variants and processed protein. In order to improve the transcription efficiency and transcript stability, codon optimization is the starting point to the chimeric creation of a gene sequence that would be introduced into a specific host organism. The codon optimization was achieved by improving codon adaptation index (CAI) which is a measurement of the relative adaptiveness of the codon usage bias for a particular DNA or RNA sequence [71], the frequency of optimal codon (FOP) used to display the optimization level of the synonymous codon choice in each gene during the translation process [72], the overall GC content of the gene and eliminating negative components that may incorporate unfavorable secondary structures on mRNA. A codon adaptation index of 0.84 was achieved by the optimized gene sequence. At its ideal, a maximal CAI design would show a value of 1.0 and a CAI of >0.8 is considered as good although no natural bacterial gene has such theoretical value [73].The percentage distribution of codons was 64%. A low GC content and disapproving peaks that could cause extremely quick mRNA turnover were also transformed and distribution was balanced to 50.33%. Furthermore, rare codons, destabilizing elements and repetitive sequences that may be detrimental to protein expression levels in E.coli BL21 [64] were eliminated. According to the energy minimization, the predicted chimeric protein would have the acceptable stability with the lowest energy (-25414.018 kcal/mol). The Mfold server was used for the prediction of RNA secondary structure. A theoretically tractable DP algorithm which is employed by the Mfold software can find the minimum ΔG structure within its thermodynamic model and the high ability to predict true positive base pairs. These results showed that the mRNA was stable enough for the efficient translation in the prokaryotic host. Five models generated by I-TASSER server for the synthetic construct and the chemical features of the modeled structure were assessed by Ramachandran plot. As described in the result section, 65.6 % resides clustered in the favored, 24.5 % in the allowed regions indicated that the backbone of the structure, φ and ψ, were reasonably accurate. Therefore, results of RAMPAGE and PROCHECK servers for the chimeric fusion protein showed that 90.1% of the residues were totally located in the favored and allowed regions and only 9.9% of the total protein were in the outlier, it is worth nothing that these negligible residues were not a part of the conformational B-cell or T-cell epitopes under investigation and could possibly be due to the presence of chimeric junctions. Consequently, it seems that outliers would not have adverse effects on the chimeric protein quality. The data of Z, C, and TM scores and RMSD for accuracy of predicted 3D models by I-TASSER and the result of model quality confirmation by the ProSA-web showed that synthetic chimeric protein has features which are the characteristics of native structures. Regarding protein structure prediction, the fusion protein designed four domains that were separated by three main coil moieties which can help the fusion protein to form a final structure. The two GS linkers which are placed between FliD and UreB domains are likely to facilitate the folding of multimeric proteins. These coil structures are associated with special amino acid sequences, residues 600-610, 668-673 and 735-740, inserted between each segment. These fragments would support the stable structure of the fusion protein which contained four domains. The effectiveness of a vaccine significantly depends on stimulating a strong immune response containing both memory B and T cells [74]. The recombinant subunit vaccines which consist of epitopes can induce a more effective response than the immunity conferred by whole protein vaccines [75]. B-cell epitopes for the chimeric protein could be predicted on the basis of the solvent accessibility, antigenicity, flexibility, hydrophilicity and secondary structure analyses [64]. Only epitopes with high prediction scores as continuous and discontinuous B-cell epitopes were selected by ABCpred and discotope servers, respectively. T-cell epitopes were selected with high potential to interact with human HLA alleles. Regarding to the solubility importance, the presence of amino acids with the high accessible value such as LYS, SER, ASP, ASN, GLY and ALA would possibly result in the chimeric protein expressed in soluble form. Consequently, all epitopes would be possibly explored to immune system in in vivo that is very important to develop of a recombinant vaccine [75].
CONCLUSION
Our data showed that the possibility of successful production of a large chimeric protein composing of four domains FliD (1-600), UreB(327-334), VacA (744-805) and CagL (51-100) in the prokaryotic host. The epitopes of the synthetic chimeric protein can induce both humoral and cellular immune responses. These findings will intensify efforts to develop a protective vaccine against H.pylori infection and may also suggest this synthetic chimeric protein could help to diagnosis of the H. pylori infection. Further studies are required to stablish these notions which are the theme of our future researches.
FINANCIAL DISCLOSURE
This work was supported by grants from Iran National Science Foundation: INSF (grant No. 93034823) and Kashan University of Medical Sciences (grant no.93194).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

