All published articles of this journal are available on ScienceDirect.

Evaluation of a Commercial Multiplex PCR for Rapid Detection of Multi Drug Resistant Gram Negative Infections
Abstract
Introduction:
Community and healthcare associated infections caused by multi-drug resistant gram negative organisms (MDR GN) represent a worldwide threat. Nucleic Acid Detection tests are becoming more common for their detection; however they can be expensive requiring specialised equipment and local expertise. This study was done to evaluate the utility of a commercial multiplex tandem (MT) PCR for detection of MDR GN.
Methods:
The study was done on stored laboratory MDR GN isolates from sterile and non-sterile specimens (n=126, out of stored 567 organisms). Laboratory validation of the MT PCR was done to evaluate sensitivity, specificity and agreement with the current phenotypic methods used in the laboratory. Amplicon sequencing was also done on selected isolates for assessing performance characteristics. Workflow and cost implications of the MT PCR were evaluated.
Results:
The sensitivity and specificity of the MT PCR were calculated to be 95% and 96.7% respectively. Agreement with the phenotypic methods was 80%. Major lack of agreement was seen in detection of AmpC beta lactamase in enterobacteriaceae and carbapenemase in non-fermenters. Agreement of the MT PCR with another multiplex PCR was found to be 87%. Amplicon sequencing confirmed the genotype detected by MT PCR in 94.2 % of cases tested. Time to result was faster for the MT PCR but cost per test was higher.
Conclusion:
This study shows that with carefully chosen targets for detection of resistance genes in MDR GN, rapid and efficient identification is possible. MT PCR was sensitive and specific and likely more accurate than phenotypic methods.
INTRODUCTION
Community and healthcare associated infections caused by multidrug resistant gram negative organisms (MDR GN) represent a major threat worldwide [1, 2]. Various resistance mechanisms exist with the most common and concerning including extended spectrum beta lactamase (ESBL), AmpC beta lactamase, carbapenemase, efflux pumps, porin mutations and other target alterations. While the knowledge of the resistance genotype of MDR GN may not impact on empirical antimicrobial treatment, it is important for infection control programs and prescription of definitive antimicrobial therapy [3-7]. Delay in identifying and reporting antimicrobial susceptibility has important clinical implications as MDR enterobacteriaceae have less favourable outcomes than non-MDR organisms [8-10].
Nucleic Acid Detection tests (NAD) are emerging as rapid and high throughput tests for detection of antimicrobial resistance. Decreasing time to result could guide rapid prescribing of definitive antimicrobial therapy and reduce the cost and burden of inappropriate antimicrobial therapy [11-13]. Clearly however, cost can be a major barrier to the establishment of molecular testing for MDR GN [11, 13]. Although many phenotypic and genotypic singleplex, multiplex, real time polymerase chain reaction (PCR), DNA microarray tests are available for categorisation of resistance in gram negative organisms, their utility will depend on the local availability of expertise and equipment and the characteristics of the patient population serviced by the laboratory [14, 15]. With these considerations in mind, we designed this study to evaluate and to assess the utility of a commercially available multiplex tandem PCR assay (MT PCR) for gram negative resistance detection. The assay could be performed in the laboratory within the existing infrastructure and resources and promised to improve the coverage and the significant delays in time to result experienced by our current phenotypic method.
MATERIALS AND METHODS
1. Study Setting
South Western Sydney Local Health District covers a large urban and semi-rural area with a population of approximately 1 million people. The Sydney South West Pathology Service Microbiology laboratory provides services to all of the 6 public hospitals of the local health district comprising over 2000 acute hospital beds. For the purposes of the study multi-drug resistant gram negatives (MDR GN) are defined as gram negative bacteria that are not susceptible to at least 1 agent in 3 antimicrobial classes [1]. MDR GN isolates from sterile sites are routinely stored for future research purposes.
Current screening for MDR GN and standard confirmatory tests for MDR GN (phenotypic identification method) are described below. The Vitek2 (bioMerieux, Marcy l’Etoile, France) MIC profile acts as a screening test that leads to phenotypic, and sometimes genotypic, confirmatory tests. For enterobacteriaceae an MIC ≥ 2 mg/L for any one of the third generation cephalosporins (3GC), or cefepime (FEP), or ceftazidime (CAZ), or a gentamicin MIC ≥ 4 mg/L, or a meropenem (MEM) MIC of ≥ 0.5 mg/L, triggers further testing. For Acinetobacter species the screening triggers are ceftazidime or cefepime MIC ≥ 4 mg/L, or meropenem MIC ≥ 2 mg/L; and for Pseudomonas species the trigger is a ceftazidime or cefepime MIC ≥ 8 mg/L, or meropenem MIC ≥ 4 mg/L. Antimicrobial susceptibility testing and phenotypic confirmation of MDR GN are currently done in our laboratory as per CLSI recommendations. The confirmatory test is a combination of double disc synergy testing (DDST) and disc approximation (DA) used in tandem on a combination of three Mueller Hinton agar (MHA) plates based on the CLSI document [16]. Based on the results of the phenotypic testing further genotypic testing for carbapenemase like IMP, VIM and SPM is performed in the laboratory, however no genotypic testing is done for ESBL and AmpC producing organisms.
a) Ethics Approval
As this study involved testing of patient isolates, ethics approval was obtained from the South Western Sydney Local Health District (SWSLHD) Human Research and Ethics Committee (SWSLHD 14/005 LNR).
a) Selection of the Targets for the MT PCR
Although the MT PCR was produced by a commercial company (AusDiagnostics Pty Ltd, Beaconsfield, NSW, Australia) the investigator was given the responsibility for choosing gene targets appropriate to the local setting. Targets were chosen to maximise the detection of ESBL and carbapenemases that would be encountered in clinical specimens. A retrospective audit over 7 years was used to determine the type of MDR GN that have been isolated in our laboratory from sterile site and surveillance swabs. This included results from a previous validation study of the phenotypic confirmatory method described above [17]. Other factors considered included two prevalence studies of MDR GN from the Sydney area and recent testing recommen-dations provided by the Australian Commission on Safety and Quality in Health Care for detection of carbapenem resistant enterobacteriaceae [18-20]. OXA23 was chosen over OXA48 because more than 12% of the MDR GN isolates isolated by the laboratory were non-fermentative bacteria such as Acinetobacter species and Pseudomonas species and OXA23 is more common in these types of organisms. The final make-up of the MT PCR is shown in Table 1.
b) Multiplex Tandem PCR (MT PCR) Method
A single isolated colony from a pure culture was picked using a 10 µl loop and suspended in 0.1 ml of Tris-EDTA buffer. Organism DNA was extracted by heating at 96°C for 10 min in a heating block. This extract was centrifuged at 12000G for 2 minutes and 10 µl of supernatant from this suspension was diluted in 1 ml of sterile water (1:100 dilution). This specimen was vortexed for 10 seconds before being utilized for PCR reactions. The first round of amplification was a short 15 cycle pre-amplification reaction using a mixture of primers homologous to each of the 11 targets and internal control. The product was transferred to individual wells containing a single specific target primer nested within those of step one (tandem PCR) [21]. The process was automated using the Easy plex liquid handling robotics system (AusDiagnostics). The second amplification was performed in the Rotor-Gene (RG6000) thermal cycler and the presence of product was detected by an increase in fluorescence of the inculcating Eva-Green dye. Fluorescence was measured at the end of each 72°C extension step and product specificity further checked by melt curve analysis.
c) Interpretation of MT PCR Results
A run was only valid when the internal control was detected at a quantification cycle (Cq) of < 20 cycles, and the melt curve of the internal control was within expected limits. The analysis software (AusDiagnostics) interpreted a target as present if the melt characteristics of the product conformed to analysis software (preset) values. Any sample with curve Cq less than 10 was further diluted (1:1000). When more than one target was detected in a single organism (such as combination of CTX-M and TEM or CMY), results were only accepted when melt curve characteristics supported the presence of all targets.
3. Workflow and Cost Analysis
Costs of reagents and labour were estimated for the standard phenotypic method and the MT PCR. The labour cost was estimated to be AU $50 per hour of work. A number of workflow scenarios were considered in order to estimate the time to result for each method.
4. Study Design
Validation Study
The validation of the MT PCR was performed in three stages.
a) Performance of the MT PCR Against Control Organisms of Known Genetic Resistance Mechanism
Twenty three reference bacterial strains were obtained from two Australian reference laboratories. These organisms had their resistance mechanism well characterised by molecular sequencing. These isolates were supplemented with eight American type culture collection (ATCC) strains with no known resistance mechanism. Initial sensitivity and specificity of the MT PCR were determined by this testing. [22]. This is shown in Table 2.
b) Performance of the MT PCR Against Organisms with Known Phenotypic Resistance
A search of laboratory records identified 567 organisms from a range of clinical and routine surveillance samples tested in the laboratory between 1st January 2008 and 28th February 2014 that had a resistance mechanism confirmed by the current phenotypic method (Fig. (1) line 1). Organisms were selected to allow approximately 50 resistance phenotypes from each group (ESBL, AmpC, and Carbapenemase) to be tested in the MT PCR. MT PCR was done on a total of 126 organisms of these 567 (Fig. (1) line 2). A total of 156 phenotypic resistance mechanisms were found in these 126 organisms as some had more than one resistance mechanism (i.e. ESBL+ AmpC or ESBL+ Carbapenemase). A sample of organisms from each group were further tested by a second nucleic acid amplification technique for the relevant resistance method (labelled “multiplex PCR” in Fig. (1) line 4) and for another small sample, sequencing of the DNA product produced by the MT PCR reaction was performed to confirm the specificity of the MT PCR reaction (labelled “sequencing” in Fig. (1) line 4).
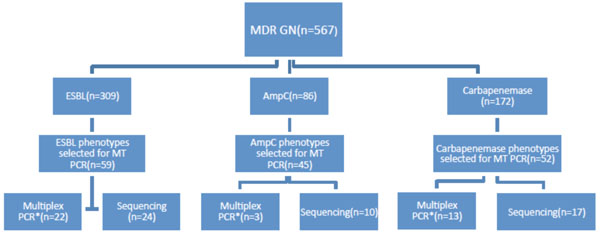
Organism selection for validation of MT PCR against current phenotypic method.
Genetic resistance targets selected for the MT PCR and the corresponding genotypes detected.
| Position Number | Phenotypic Mechanism Detected | Abbreviated Molecular Target Genes | Genotypes Detected |
|---|---|---|---|
| 1 | ESBL | pan-TEM | blaTEM types (1, 104-106, 71, 76-84, 138, 143, 150, 155). |
| 2 | ESBL | pan-SHV | blaSHV types (1, 2, 11, 25, 26, 38, 56) |
| 3 | ESBL | CTX-M group 1 | blaCTX-M-1 (1, 3, 15, 28, 29, 32, 36, 58, 79, and 103) |
| 4 | ESBL | CTX-M group 9 | blaCTX-M-9 (9, 13, 14, 24, 27, and 38) |
| 5 | AmpC | DHA-1 | blaDHA beta lactamases from a range of enterobacteriaciae species |
| 6 | AmpC | pan-CMY | blaCMY (1, 10, 11, 8b, 19) |
| 7 | Carbapenemase | pan-VIM | blaVIM (1,2, and 3) |
| 8 | Carbapenemase | pan-IMP | blaIMP (1, 4, 5, 6, 10) |
| 9 | Carbapenemase | OXA-23 | blaOXA-23 |
| 10 | Carbapenemase | KPC | blaKPC (1, 2, 3) from Klebsiella and other bacterial species including Pseudomonas and E. coli. |
| 11 | Carbapenemase | NDM | blaNDM (1,2,3) |
| 12 | Nil | Artificial sequence for assay control | Spike |
Reference bacterial strains and MT PCR results.
| No. | Strain | Organism | Resistance Gene | Primary Gene Detected by MT PCR | Source or Reference |
|---|---|---|---|---|---|
| 1 | JIE142 | K. pneumoniae | blaSHV-11 | SHV | Thomas and Olma |
| 2 | PA185 | P. aeruginosa | blaSHV-5 | SHV | [18] |
| 3 | JIE181 | K. pneumoniae | blaSHV-28 | SHV | Thomas and Olma |
| 4 | JIE058 | E. coli | blaCTX-M-27(M9 group) | CTX-M-9 | [18] |
| 5 | JIE88 | E.coli | blaCTX-M-14(M9 group) | CTX-M-9 | [18] |
| 6 | JIE251 | E.coli | blaCTX-M-3(M1 group) | CTX-M-1 | [18] |
| 7 | NS249 | S. marcescens | blaIMP-11 | Not Detected | Turnidge |
| 8 | JIE137 | K. pneumoniae | blaCTX-M-62(M1 group) | CTX-M-1 | [18] |
| 9 | JIE162 | K. pneumoniae | blaCTX-M-15/SHV12(M1 group) | CTX-M-1/SHV | [18] |
| 10 | N6994 | P. aeruginosa | blaIMP-1 | IMP | Turnidge |
| 11 | JIE203 | K. pneumoniae | blaDHA-1 | DHA-1 | Thomas and Olma |
| 12 | JIE298 | E. coli | blaCTX-M-24(M9 group) | CTX-M-9 | [18] |
| 13 | JIE144 | P. aeruginosa | blaIMP-7 | IMP | Turnidge |
| 14 | N12636 | P. aeruginosa | blaVIM-3 | VIM | Turnidge |
| 15 | JIE602 | E.coli | blaCMY-2 | CMY | Thomas and Olma |
| 16 | 09K280459L | E.coli | blaNDM-1 | NDM-1 | Taylor, [19] |
| 17 | KPN2303 | K. pneumoniae | blaKPC-2 | KPC | Quinn, [19] |
| 18 | OXA Org | E.coli | blaOXA -23 | OXA23 | [19] |
| 19 | OXA Org | K. pneumoniae | blaOXA-48 | Not Detected | [19] |
| 20 | NDM1 | K.pneumoniae | blaNDM-1 | NDM-1 | Sidjabat and Paterson |
| 21 | OXA48 | K. pneumoniae | blaOXA-48 | Not Detected | Sidjabat and Paterson |
| 22 | OXA23 | A. baumanii | blaOXA-23 | OXA 23 | Sidjabat and Paterson |
| 23 | KPC | K. pneumoniae | blaKPC | KPC | Sidjabat and Paterson |
| 24 | ATCC 25922 | E. coli | nil | Not Detected | [22] |
| 25 | ATCC 35659 | P. mirabilis | nil | Not Detected | [22] |
| 26 | ATCC 13883 | K. pneumoniae | nil | SHV | [22] |
| 27 | ATCC 27592 | S. liquefaciens | nil | Not Detected | [22] |
| 28 | ATCC 13047 | E. cloacae | nil | Not Detected | [22] |
| 29 | ATCC 19606 | A. baumanii | nil | Not Detected | [22] |
| 30 | ATCC 43863 | K. oxytoca | nil | Not Detected | [22] |
| 31 | ATCC 27853 | P. aeruginosa | nil | Not Detected | [22] |
Note- SHV detected in K. pneumoniae was deemed as chromosomal in origin, K. oxytoca do not have SHV, and instead have OXY1/2 which code for ampicillin resistance.
Source of bacterial isolates- Thomas and Olma, Lee Thomas and Tom Olma, ICPMR, Westmead Hospital, Sydney, Australia; Bell and Turnidge, Jan Bell and John Turnidge, Women’s and Children’s Hospital, Adelaide, Australia; Taylor, Peter Taylor, Prince of Wales Hospital, Sydney, Australia; Quinn, J. Quinn, Chicago Infectious Disease Research Institute, Chicago, IL; Sidjabat and Paterson, Hanna Sidjabat and David Paterson, The University of Queensland, UQ Centre for Clinical Research, Queensland, Royal Brisbane and Women's Hospital Campus, Brisbane, Australia.
Validation against Phenotype: agreement of MT PCR with phenotypic testing.
| Phenotype | Organism Total (n=126) | MT PCR results | ||||||
|---|---|---|---|---|---|---|---|---|
| ESBL (Genotype) | AmpC (Genotype) | Carbapenemase (Genotype) | Nil Detected | |||||
| ESBL only | 32 | 30 CTXM-1-18 CTXM-9-8 TEM-3 SHV-1 |
0 | 1 IMP-1 |
1 | |||
| ESBL plus AmpC | 15 | 13 CTXM-1-9 CTXM-9-2 TEM-2 |
1 DHA-1 |
0 | 1 | |||
| ESBL plus carbapenemase | 12 | 12 CTXM-1-7 CTXM-9-3 TEM-2 |
0 | 12 IMP-12 |
0 | |||
| AmpC only | 27 | 10 CTXM-1-5 CTXM-9-2 TEM-2 SHV-1 |
9 CMY-9 |
0 | 8 | |||
| AmpC plus carbapenemase | 3 | 0 | 3 CMY-3 |
3 IMP-3 |
0 | |||
| Enterobacteriaceae producing carbapenemase only | 21 | 0 | 0 | 21 IMP-19 VIM-2 |
0 | |||
| Non Fermenters producing Carbapenemase only | 16 | 0 | 0 | 1 VIM-1 |
15 | |||
| % Agreement MT PCR and phenotype | Agreement value (%) | |||||||
| ESBL overall | 59 | 93.2 | ||||||
| AmpC overall | 45 | 28.8 | ||||||
| Carbapenemase | 52 | 71.1 | ||||||
Comparison of MT PCR to a second multiplex PCR for resistance detection.
| MT PCR Result | Second Multiplex PCR Concordant | Second Multiplex PCR Discordant | Examples of Difference | Total |
|---|---|---|---|---|
| CTX-M-1 | 8 | 2 | One CTX-M-9 in multiplex PCR, one nil detected | 10 |
| CTX-M-9 | 3 | 1 | CTX-M-1 detected | 4 |
| TEM | 0 | 4 | TEM target not present in second multiplex PCR | 4* |
| SHV | 7 | 1 | 1 SHV not detected | 8 |
| CMY | 2 | 0 | No discrepancy | 2 |
| DHA | 1 | 0 | No discrepancy | 1 |
| IMP | 10 | 1 | 1 IMP not detected by multiplex PCR | 11 |
| VIM | 2 | 0 | No discrepancy | 2 |
* - excluded from analysis
Confirmation of MT PCR amplicon product
| Amplicon Types | Total Sequenced | Correct | Incorrect | Poor Sequence Detected |
|---|---|---|---|---|
| TEM | 5 | 4 | 1 | 0 |
| SHV | 5 | 5 | 0 | |
| CTXM-1 | 8 | 6 | 2 | |
| CTXM-9 | 6 | 4 | 2 | |
| CMY | 6 | 4 | 2 | |
| DHA | 4 | 1 | 1 | 2 |
| IMP | 5 | 1 | 4 | |
| VIM | 3 | 2 | 1 | |
| OXA-23 | 3 | 1 | 2 | |
| NDM | 4 | 4 | 0 | |
| KPC | 2 | 1 | 1 | |
| Total | 51 | 33 | 2 | 16 |
A significant number of organisms met the screening criteria for MDR GN but the phenotypic resistance pattern was not found on phenotypic testing. A small group (n=12) of these organisms that met MIC screening criteria for MDR GN, but for whom the phenotypic method failed to confirm the presence of ESBL, AmpC or carbapenemase, were also tested by MT PCR.
For the purposes of this part of the study organisms stored at -80°C were passaged twice on horse blood agar. To confirm the stability of the phenotypic resistance and the storage process a randomly selected group of 50 organisms selected from all three resistance groups underwent re-identification and susceptibility screening by Matrix assisted laser desorption ionization time of flight mass spectrometry (MALDITOF MS) and Vitek2 and were then retested by the phenotypic resistance testing method. This process confirmed the stored identity of the organisms in all cases and therefore MT PCR was performed on the other isolates without prior reconfirmation of phenotype. The second multiplex PCR testing for ESBL, AmpC and MBL was performed in a reference laboratory [19]. The sequencing of the MT PCR amplicons was performed on an ABI3730XL sequencer with the results compared to sequences stored in GenBank using BLAST program.
c) Limit of Detection (LOD)
This was attempted using reference strains 8, 11 and 16 as in Table 2. These strains represented organism from each different genotypic categories (CTXM-1, DHA-1 and NDM-1). Pure colonies of these organisms were obtained from culture plates and 0.5 McFarland suspension was made representing 1.5 × 108 c.f.u/ml (colony forming units/ millilitre). Serial dilutions were performed in PBS to a final dilution of 1.5 × 10-2 c.f.u/ml. These dilutions were tested in the same way as described in MT PCR method for determination of LOD.
RESULTS
1. Validation Study – Performance of MT PCR Against a Panel of Reference Strains of Known Resistance Mechanism
Results of the MT PCR testing of 31 reference strains are shown in Table 2.
The MT PCR detected 20 out of 21 expected resistance mechanisms correctly. The pan-IMP carbapenemase assay of the MT PCR failed to detect the blaIMP11 of strain 7. The two OXA 48 reference strains were not detected by the MT PCR. This was expected because the MT PCR does not contain primers for the OXA 48 gene and therefore this does not represent a failure of the assay. This analysis revealed a good overall analytical sensitivity of the MT PCR with 95% (20/21) of genetic mechanisms detected.
The MT PCR demonstrated high analytical specificity too. All ATCC strains without resistance mechanisms were negative except for strain 26 in which SHV was detected. SHV is an intrinsic, narrow spectrum, chromosomally encoded beta lactamase that is present in Klebsiella species and this was detected in all K.pneumoniae species in the validation study (strain numbers 1, 3, 8, 9, 11, 17, 19, 21, 23, 26). The pan-SHV target of the MT PCR cannot distinguish between chromosomal and plasmid derived SHV. The specificity of the MT PCR was therefore 96.7 % (30/31).
2. Performance of MT PCR Against Organisms with a Resistance Phenotype Detected by Routine Laboratory Methods
There were 156 phenotypic resistance mechanisms identified in 126 organisms (some organisms displayed more than one phenotypic resistance mechanism e.g. ESBL plus carbapenemase). The MT PCR detected 131 out of the 156 resistance mechanisms (83.9%). The MT PCR agreed with the phenotypic testing for 101 of the 126 organisms (80%). The major lack of agreement was noted in the detection of AmpC and the detection of carbapenemase in the non-fermentative organisms (see Table 3).
The MT PCR detected ESBL in 55 of the 59 phenotypic ESBLs. Interestingly for one organism that tested phenotypically as ESBL alone, a carbapenemase (metallo-beta lactamase - MBL) was detected by MT PCR. The MT PCR also detected ESBL in 10 organisms that had been phenotypically characterised as AmpC producers only. All ESBL plus carbapenemase combinations were correctly identified by the MT PCR. MT PCR performed poorly for organisms that were phenotypically characterised as having AmpC with agreement of only 28.8% (13/45). MT PCR missed 18 out of 27 organisms phenotypically characterised as AmpC alone and 14 out of 15 AmpC when in combination with ESBL. The MT PCR performed well in the detection of carbapenemase in Enterobacteriaceae detecting all of the carbapenemase-alone phenotypes and all carbapenemase-combination phenotypes (overall 71.1%, 37/52). However, the MT PCR failed to detect carbapenemase genes in the majority of non-fermentative organisms with phenotypic tests suggestive of carbapenemase production. A single Pseudomonas species harbouring blaVIM was detected, but the other 15 were not. Two Pseudomonas isolates proven to carry SPM and GES genes were not detected by the MT PCR. This is not a failure as the MT PCR does not include primers for SPM and GES targets. The breakdown of the genotypic mechanism of resistance seen in each individual phenotypic category is shown in Table 3.
a) Agreement Between MT PCR and Another Multiplex PCR
A subgroup of the GN organisms with phenotypic resistance (n=38) underwent testing by a second, different multiplex PCR. Agreement between the MT PCR and the second multiplex PCR was good (33/38 – 87% agreement) (Table 4). In this table the examples of difference are stated. The second multiplex PCR did not include targets for TEM ESBL and so these organisms were excluded from analysis.
b) Confirmation of MT PCR Amplicon Specificity by DNA Sequencing
A total of 51 organisms with phenotypic resistance had sequencing of the MT PCR amplicon performed. In 33/35 cases the MT PCR amplicon sequence aligned with a published Genbank sequence for the expected resistance gene (Table 5) 2 sequences, one TEM which did not have a sequence assigned and one having DHA which was detected as a CTXM-1. 16/51 could not have a sequence matched to them which was thought to be due to background interfe-rence or very late amplification.
c) MT PCR Results for Organisms that Meet MIC Screening Criteria for Possible MDR GN but for Whom the Phenotypic Disc Diffusion Testing Fails to Identify a Resistance Mechanism
MT PCR was performed on 12 isolates which did not have a phenotypic resistance mechanism detected by the current disc diffusion method despite presence of MIC screening criteria (based on Vitek2 screening results). MT PCR detected TEM ESBL genes in 9 organisms and CTX-M-1 ESBL gene in 1 organism. Only 2 organisms of the group were negative by MT PCR (Data not shown).
d) Limit of Detection
The MT PCR accurately identified all the resistance genotypes in all serial suspensions up to dilution of 1.5 × 102 c.f.u/ml. No amplicon product was detected below this dilution.
a) Phenotypic Testing
The cost of agar plates and antibiotic discs for the current phenotypic method is approximately AUD $6 per organism. The hands on time for the set up and interpretation is approximately 30 minutes per organism and therefore the cost in labour is AU $25 per organism added to this. Hence total cost of phenotypic test is AU $31/organism. If the phenotypic testing suggests carbapenemase is present, an in-house PCR test is performed and this costs AU $10 per organism for reagents and AU $50 per organism for labour, in which case the cost of testing would increase to AU $91/organism. The time to result (turnaround time for phenotypic results) for the phenotypic test is approximately 24 to 36 hours after the Vitek2 results are available for ESBL and Amp C and 36 to 48 hours after Vitek2 results for carbapenemase. There is no limit to batch size on phenotypic testing and batch size does not affect the time to result for this test. Some organisms with phenotypic carbapenemase profile but negative in the in-house PCR are sent to a reference laboratory for further testing with substantial delay and added cost.
b) MT PCR
The reagent and the kit cost for the MT PCR is AU $30 per organism. The hand on time for set up and interpretation is approximately AU $25 per organism and therefore total cost of MT PCR is AU $55/organism. The time to result of the MT PCR assay is 3 hours after the Vitek2 result. There are equipment requirements for the MT PCR and this is likely to be more than AU $50,000 establishment cost. Our laboratory performs other MT PCR assays and so there was no equipment cost for our lab. The broader range of the MT PCR assay may reduce the necessity for referring samples to another lab for confirmation.
The batch size will affect the cost and the time to result for the MT PCR. A batch of 6 specimens can be tested in a single run. Decreasing the number of organisms tested in the run will increase the cost per organism due to wastage and costs of controls. Alternatively waiting for a full batch could significantly delay the results. It is estimated that the laboratory currently performs 10 to 12 confirmatory phenotypic assays per week and therefore the time to result for full batch testing under current testing algorithms will be approximately 3 to 3.5 days. A smaller batch size such as 3 per batch could support daily testing at an increased cost (AU $63/organism).
DISCUSSION
The MT PCR performed well when validated against organisms with known genetic mechanism of resistance. The sensitivity of the MT PCR was 95%, only missing one IMP carbapenemase and two OXA 48 carbapenemase, although the MT PCR did not have targets for OXA-48 detection. The only false positive was the detection of SHV in a Klebsiella pneumoniae (ATCC strain) which could be due to the intrinsic (chromosomal) production of SHV [23]. Overall specificity of the PCR was found to be 96.7%. These results are comparable with studies of other multiplex assays for gram negative resistance detection [24, 25]. In a study of 489 gram negative isolates which were tested by a commercially available PCR, ESBL was detected with a sensitivity of 98.3% and a specificity of 100% [26]. However in that study, DNA microarray was the gold standard used, not a PCR based assay as in our study. In another study, a commercially available PCR for carbapenemase performed with a sensitivity of 96.7% and specificity of 99% when tested against 132 enterobacteriaceae [27]. Detection of 3 blaIMP harbouring strains was missed by this assay and this was thought to be due to diversity within the IMP family. This problem was also found in our evaluation of the MT PCR which failed to detect a case of blaIMP. This could be because the pan-IMP primers did not cover the IMP 11 gene or it could be due to individual strain polymorphism.
When MT PCR performance was compared to phenotypic methods of detection of resistance, the agreement was much lower. Although the detection of ESBL was good (93.3%), the detection of AmpC betalactamase in all organisms and the detection of carbapenemase in non-fermentative organisms was poor (29% and 6% respectively). There could be a number of reasons for the poor agreement for AmpC detection. Firstly as only two AmpC targets (CMY-1 and DHA) were chosen in the MT PCR assay, this could have limited the detection of other types of AmpC betalactamase. Secondly these two targets may not be the prevalent AmpC in our population. Although there is only limited data about the epidemiology of the prevailing AmpC types, the two AmpC targets were chosen based on a previous study from our laboratory and so they should have performed fairly well [17]. Another possible explanation would be poor specificity of the phenotypic AmpC detection method. There appears to be particular problem with the detection of AmpC when the organism also produces ESBL. It has been shown that clavulanic acid may induce a high level expression of chromosomal AmpC enzyme and this may antagonize the activity of the partner beta lactam which in turn leads to masking of synergy required to detect ESBL [28]. This could explain some of the phenotypic testing reported as AmpC production instead of ESBL production.
The MT PCR results for the detection of carbapenemase demonstrated much higher agreement for the enterobacteriaceae (100%) than for the non-fermentative organisms (6%). It is known that non-fermentative organisms (such as Pseudomonas species) can have multiple mechanisms conferring phenotypic carbapenem resistance including altered membrane permeability, efflux pumps and other enzymes and therefore such phenotypic resistance may not be due to a carbapenemase gene [29]. This is supported by our findings in another study that 39 of 40 Pseudomonas aeruginosa isolates with apparent carbapenemase on phenotypic testing were negative for common metallo-betalactamase genes (IMP, VIM, SPM, GIM, SIM) on molecular testing. It was thought that the intrinsic antibacterial activity of the EDTA contributed to these false positive phenotypic results for Pseudomonas species [30]. Alternatively, a novel and as yet undefined carbapenemase could be present in these organisms and this would not be detected by the MT PCR assay. In other studies evaluating carbapenemase production in Pseudomonas aeruginosa have found intra hospital clonality (when tested from single centres) or poor sensitivity of genotypic tests [31, 32]. It is possible therefore that the results of the MT PCR detection of carbapenemase in non-fermenters are accurate and it is the phenotypic testing that is misleading/suboptimal. Optimal methods for phenotypic testing for non-fermenters needs to be further studied.
Various phenotypic methods for ESBL detection have a varying sensitivity and specificity in literature. This depends on the indicator antibiotic used, principle of the test (double disc synergy vs. disc approximation) and various combinations of indicator/inducer combinations that are tested. In the literature the sensitivity of combined phenotypic methods range from 85-100%, however they have lower specificity ranges from 75-98% [33-36]. When we tested 12 enterobacteriaceae that met Vitek2 MIC screening criteria for multi-resistance, but for which phenotypic methods had failed to detect any resistance phenotype, the MT PCR found ESBL genes in 83%. This could be explained by under-expression of these ESBL genes leading to false negative results in phenotypic tests. It is well known that detection of ESBL phenotypes can be missed when only a single method is used (such as double disc synergy or E-test alone), or when a single antibiotic substrate is used (such as cefpodoxime or cefotaxime alone) [35, 36].
The MT PCR demonstrated some substantial benefits over the current phenotypic method for confirming and defining the resistance mechanism for gram negative organisms. The MT PCR could reduce the time to result by approximately 2 days. This would allow clinicians to make treatment decisions earlier potentially reducing time to definitive antimicrobial therapy and to institution of appropriate infection prevention and control measures. In another study done in our hospital, we found that inappropriate antimicrobial therapy is associated with increased mortality in ESBL producing enterobacteriaceae bacteraemia, and so this reduction in time to result may have outcome benefits for the patient, although this still needs to be tested [37].
However, as with any rapid nucleic acid amplification test, the cost of the test and the requirement for suitable expertise can limit the time benefits, as the tests need to be run in batches to make them economically viable [12, 24]. This is true also of the MT PCR in the format that we tested. Factors such as the local prevalence of MDR GN, the resources available in the laboratory and the number of samples to be tested will influence at what point in the testing algorithm molecular tests would have a role [38-41]. We believe that for our laboratory, MT PCR could have a role in quickly confirming the presence of a genetic resistance mechanism when the Vitek2 screening test suggests multi-drug resistance. That is, replacing the current culture based phenotypic method. However this would only be viable if small batch testing (3 isolates) were possible and economically affordable otherwise delays in time to result would ensue. Using the MT PCR to characterise MDR GN organisms causing non-sterile site infections, and to quickly confirm genotype in infection control surveillance isolates could minimise wastage of reagents and speed up time to result. The cost of such a proposal would still need to be measured. It may also be possible to use only part of the testing disc or a modified 8-plex assay, which could save reagents and decrease the need for large batch testing (personal communication from AusDiagnostics).
It may also be possible to improve the effectiveness of the MT PCR by redesigning the assay to suit local conditions or to keep up with changes in local epidemiology of MDR GNs [19]. The MT PCR assay used in our study included gene targets for the commonly found TEM, SHV and CTX-M-1 and 9 ESBL families as well as IMP and VIM carbapenemases which are locally prevalent in Australia [3, 41]. We feel that the apparent poor performance of the AmpC targets, as well as questions about the clinical importance of detecting AmpC and some of the narrow spectrum beta lactamases such as TEM and SHV and the OXA-23 in non-fermentative organisms, warrants modification of the assay for our local use. We propose to try a new version of the MT PCR with a slightly different range of targets such as NDM, KPC, IMP, VIM, OXA 48 and most prevalent ESBL’s such as CTXM-1 and CTXM-9. These changes may also increase the number of isolates able to be tested in a single run, thereby minimising reagent wastage. Any laboratories considering using the MT PCR should carefully consider these issues and try to design the most suitable assay for local circumstances.
Regardless of how frequently the MT PCR assay is performed, it will be more expensive in reagents than the phenotypic method. Improvements in sensitivity and specificity and time to result may justify the cost, if better clinical outcomes result. Clinical outcomes such as empirical antimicrobial therapy prescribed, antimicrobial switch to definitive therapy, implications in infection control and infection related mortality need to be measured in a prospectively designed study. Reductions in referral of organisms to other labs may also off-set increased assay costs. Not all laboratory testing decisions are based on cost, jurisdictional guidelines and regulatory requirements can also affect the design of laboratory testing algorithms for MDR GNs. For example, although current CLSI guidelines (M100-S24) do not mandate the confirmation of genetic resistance mechanisms in all cases, EUCAST now considers it mandatory for epidemiological and infection control purposes [16, 40]. It is also likely that increasing efforts to control antimicrobial resistance will mandate more accurate and timely confirmation of resistance genotypes [41].
There were several limitations to our study. Firstly the number of isolates in the validation study and the number that underwent genetic sequencing to confirm the specificity of the amplicon were limited by availability and cost. It is possible that if we had included all isolates that were positive for MDR GN by screening method (Vitek2) then we could have detected additional resistance mechanisms. However current laboratory practice does not store these isolates for further testing and so they were not available for the study. The design of the MT PCR was based on local prevalence data and also regional and universal patterns of genetic resistance. The choice of gene targets is limited and may not have included other prevalent resistance genes that are either at present unknown or that are on the increase. The comparison of the MT PCR with the current phenotypic test was not performed in real laboratory conditions in this retrospective study and so the benefits, cost and limitations of each method may not reflect a real life situation.
CONCLUSION
In conclusion, this study shows that with carefully chosen targets for the detection of resistance genes in MDR GN, efficient and rapid detection is possible. The MT PCR was sensitive and specific and is likely to be more accurate than current phenotypic methods.
CONFLICT OF INTEREST
None related to this study. This study was funded internally from the department research funds.
ACKNOWLEDGEMENTS
I would like to thank the staff in the Molecular section of the microbiology laboratory for providing initial technical support during the study. Also would like to thank Dr. Lee Smith and Dr. Keith Stanley (AusDiagnostics) for their technical assistance with the MT PCR and assistance with molecular sequence analysis of the wild type isolates and Professor Jon Iredell (CIDM, Westmead) and Professor David Paterson (UQ, Brisbane) for providing the reference isolates for MT PCR.
AUTHORS CONTRIBUTION
Design of the MT PCR targets, literature review of MDR GN, business case for acquisition of the MT PCR kits, local ethics committee application, conducting all three phases of the study, data analysis and draft of the manuscript were all done by Ruchir Chavada, Michael Maley provided critical input into the data analysis and reviewed the subsequent drafts of the manuscript.

