All published articles of this journal are available on ScienceDirect.

Engineering Vibrio fischeri for Inducible Gene Expression
Abstract
The marine bacterium Vibrio fischeri serves as a model organism for a variety of natural phenomena, including symbiotic host colonization. The ease with which the V. fischeri genome can be manipulated contributes greatly to our ability to identify the factors involved in these phenomena. Here, we have adapted genetic tools for use in V. fischeri to promote our ability to conditionally control the expression of genes of interest. Specifically, we modified the commonly used mini-Tn5 transposon to contain an outward-facing, LacI-repressible/IPTG-inducible promoter, and inserted the lacI gene into the V. fischeri chromosome. Used together, these tools permit the identification and induction of genes that control specific phenotypes. To validate this approach, we identified IPTG-controllable motility mutants. We anticipate that the ability to randomly insert an inducible promoter into the genome of V. fischeri will advance our understanding of various aspects of the physiology of this microbe.
INTRODUCTION
Vibrio fischeri is a marine bacterium used as a model organism for studies of bioluminescence [1, 2], quorum sensing [3], biofilm formation [4, 5], and symbiosis [6, 7]. The discovery and characterization of these natural phenomena have been greatly facilitated by the development and use of genetic tools that disrupt and modify genes within the V. fischeri genome.
While the bioluminescent nature of V. fischeri was first described in the 19th century by Martinus Willem Biejerinck [8], it was not until 1992 when the first genetic manipulation was described by Dunlap and Kuo [9]. These researchers successfully introduced foreign DNA via conjugation and generated gene replacement mutants via homologous recombination. Specifically, they manipulated a plasmid-borne copy of the V. fischeri luminescence (lux) genes in Escherichia coli to obtain insertion and deletion mutants, then transferred the resulting constructs into V. fischeri, selecting for lux mutant recombinants. In 1994, the technique of transposon mutagenesis was applied to V. fischeri [10], thus permitting the investigation of uncharacterized genes and their involvement in phenotypes such as host colonization.
In subsequent years, the ability to manipulate V. fischeri genetically was facilitated by the identification of antibiotic resistance cassettes that functioned in this organism and, in concert, the construction and/or use of a variety of plasmids: those that were more stable were used for complementation, while those that were less stable were used for recombination [11]. Importantly, the identification of a V. fischeri-specific origin of replication facilitated the construction of highly stable vectors that (1) could be maintained without antibiotic selection and (2) were compatible with other commonly used plasmids [12], allowing experiments that depended on the use of two plasmids [13]. In addition, constructs used for inserting genes in single copy at the Tn7 site of other bacteria [14] were successfully adapted for V. fischeri [11, 15], permitting single copy gene expression and complementation in the absence of antibiotic selection and making it possible to conduct well-controlled gain of function experiments in the context of animal colonization [16-19].
The whole genome of V. fischeri was sequenced and annotated in 2005 [20], allowing researchers to more readily utilize reverse genetics approaches. Facilitating such approaches, a counter-selectable suicide vector used to generate unmarked gene replacements in Vibrio splendidus [21] was adapted for use in V. fischeri. [17, 22]. This approach has permitted the reliable and rapid construction of unmarked mutations and the ability to readily generate strains with multiple unmarked mutations.
Although V. fischeri genetics has advanced greatly since 1992, some aspects remain under-developed. For example, little work has been done to develop inducible control over the expression of genes present in the chromosome. Although plasmid libraries have been developed [23, 24] and can be used to identify positive regulators of a given phenotype, such libraries are limited in the size of the insert and are unlikely to contain large operons intact. Here, we discuss the adaptation and use of a genetic tool for V. fischeri that allows for gene disruption as well as the insertion of a conditional promoter randomly into the genome, and can thus facilitate the identification of genes, such as positive regulators or large operons, whose induction drives a desired phenotype.
MATERIALS AND METHODS
Strains and Media
The V. fischeri strains and plasmids constructed and/or used in this study are listed in Table 1. V. fischeri strain ES114 was used as the wild-type strain in these studies. V. fischeri strains were grown in complex LB-salt (LBS) medium [25] and tryptone broth-seawater (TB-SW) motility medium [26]. We also used the E. coli strains GT115 (Invivogen, San Diego, CA), CC118 [27], and π3813 [21] for the purposes of cloning and conjugation. E. coli strains were grown using either LB [28] or Brain-heart infusion (BHI) (Difco) medium. The following antibiotics were added, as appropriate, at the indicated final concentrations: chloramphenicol (Cm) at 1 to 5 µg/ml for V. fischeri and 25 µg/ml for E. coli; tetracycline (Tc) at 5 µg/ml for V. fischeri and 15 µg/ml for E. coli; kanamycin (Kan) at 50 µg/ml; erythromycin (Erm) at 5 µg/ml for V. fischeri and 150 µg/ml for E. coli; and ampicillin (Ap) at 100 µg/ml. Thymidine was added to a final concentration of 300 µM for the growth of π3813 E. coli cells. Agar was added at a final concentration of 1.5% for solid media and 0.225% for the motility medium.
Bacterial strains and plasmids.
| Strain | Genotype | Reference |
|---|---|---|
| V. fischeri | ||
| ES114 | Wild-Type | [42] |
| KV6576 | IG (yeiR-glmS)::lacIq | This Study |
| KV7432 | IG (yeiR-glmS)::lacIq IG (VF_A0340-VF_A0341)::Tn5P (Ermr) | This Study |
| KV7433 | IG (yeiR-glmS)::lacIq cheZ::Tn5P (Ermr) | This Study |
| Plasmid | Description | Reference |
| pCA24N | lacIq (Cmr) | [43] |
| pCLD46 | LacI-repressible RscS overexpression plasmid (Cmr) | [13] |
| pEVS104 | Conjugal helper plasmid (Kanr) | [32] |
| pEVS107 | Mini-Tn7 delivery plasmid (Kanr Ermr) | [15] |
| pEVS170 | Tn5 carrier plasmid (Kanr Ermr) | [31] |
| pJET1.2 | Commercial cloning vector (Ampr) | Fermentas |
| pJMO8 | pKV363 containing the yeiR-glmS IG region (Cmr) | This Study |
| pJMO10 | pEVS170 containing a promoter within the Tn (Tn5P) (Kanr Ermr) | This Study |
| pJMO14 | pJMO8 containing the lacIq allele (Cmr) | This Study |
| pKV363 | Mobilizable suicide vector (Cmr) | [22] |
| pVSV105 | Mobilizable vector (Cmr) | [12] |
Oligonucleotides.
| Primer # | Name | Sequence |
|---|---|---|
| 908 | 170Ext2 | GCACTGAGAAGCCCTTAGAGCC |
| 974 | Tn7_primerdma89 | GCTAAAGCGGTGACGGTGGAGTAG |
| 975 | Tn7_primerdma90 | CCTCACCCCAGATGGTTTGGCA |
| 1439 | Tn5-P_out Spe/Apa-F | CTAGTTTATCAAAAAGAGTGTTGACTTGTGAGCGGATAACAATGATACTTA GATTCAATTGTGAGCGGATAACAATTTCACACAGGGCC |
| 1440 | Tn5-P_out Spe/Apa-R | CTGTGTGAAATTGTTATCCGCTCACAATTGAATCTAAGTATCATTGTTATC CGCTCACAAGTCAACACTCTTTTTGATAAA |
| 1484 | VF2371 F | CTTGATTTATACAGCGAAGG |
| 1485 | VF2371 Not I R | TAGGCGGCCGCACTTAGTATGGTTTTGAAGAGTAATTAATGTTTATTG |
| 1486 | VF2372 Not I F | CATACTAAGTGCGGCCGCCTATATTGTCTCTCTTAGAACAATTATTC |
| 1487 | VF2372 R | GGTCGTGGGGAGTTTTATCC |
| 1489 | LacINotI R | GCGGCCGCCGCTCACTGCCCGCTTTCC |
| 1544 | LacIqNotI F | GCGGCCGCGACACCATCGAATGGTGCAAAAC |
Plasmid and strain construction. Standard molecular biology, transformation, and conjugation approaches were used to generate the plasmids and strains used in this study. The primers used are shown in Table 2. Plasmid pJMO14 was used to insert the lacIqgene into the chromosome between yeiR and glmS, adjacent to the attTn7 site (Fig. 2). To generate pJMO14, upstream and downstream sequences (~500 bp) flanking the target insertion site were first amplified from the ES114 chromosome by PCR using primers 1484 and 1485 and primers 1486 and 1487, then joined using overlap extension PCR [22, 29]. The resulting DNA fragment, which consisted of the flanking sequences joined by non-native sequences including a NotI site, was ligated into suicide vector pKV363 to generate pJMO8. Finally, the lacIq gene, which was amplified using PCR with pCA24N as a template and primers 1544 and1489, was cloned into the PCR cloning vector pJET1.2, then sub-cloned into NotI-digested pJMO8 to generate pJMO14. To insert lacIq into the chromosome, the method of Le Roux et al. [21] was used as described previously [22]. The insertion of lacIq into ES114, generating KV6576, was confirmed using PCR with primers 974 and 975.
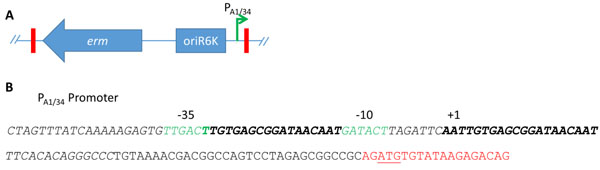
Construction of the Tn5P transposon. A) The Tn5 from pEVS170 contains an erythromycin resistance gene and an origin of replication (blue arrow and box) within the Tn5 ends (red rectangles). pEVS170 was modified to contain an outward-facing, LacI-repressible/IPTG-inducible promoter, PA1/34 (green arrow) [1]. (B) The PA1/34 promoter region (italics) contains two LacI binding sites (bold). The binding of LacI to these sites hinders access to the -35 and -10 sites (green), thus repressing transcription. The Tn5 end (red) contains a start codon (underlined).
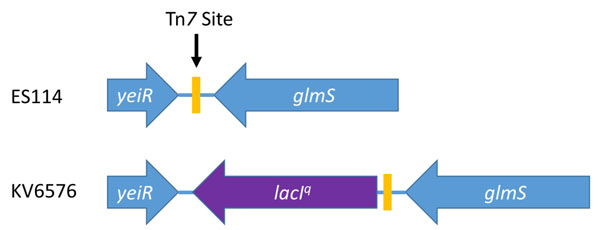
Construction of a lacI-expressing V. fischeri strain. ES114 contains a Tn7 site (yellow rectangle) between yeiR and glmS. KV6576 was engineered to contain the lacI overexpression allele (lacIq, purple arrow) near, but not disrupting, the Tn7 site.
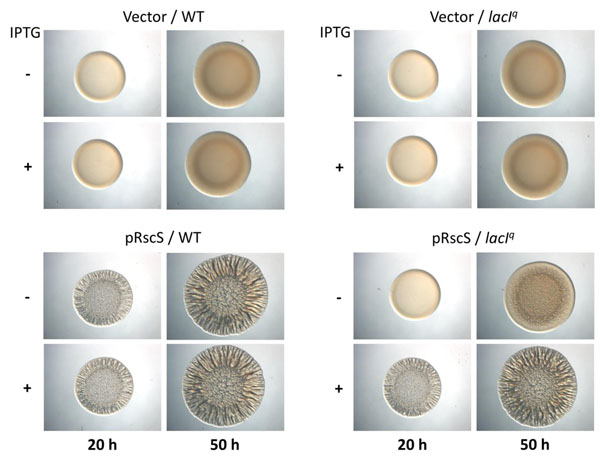
lac promoter-driven biofilm formation by WT and lacI expressing strains. Cultures of wild-type (ES114) and lacIq (KV6576) strains containing either the empty vector pVSV105 or RscS expression plasmid pCLD46 were grown in LBS containing Cm. Aliquots were diluted to an OD600 of 0.2, spotted onto LBS-Cm medium containing or lacking 1.75 mM IPTG, and incubated at room temperature. Wrinkled colony formation was assessed at 20 and 50 h post inoculation.

Migration of mutant strains on soft agar. KV6576 (A), KV7432 (B), and KV7433 (C) were grown overnight in LBS. Cultures were diluted to an OD600 of 0.4 prior to inoculation on TB-SW motility medium containing or lacking 1.75 mM IPTG. The images depict migration after 5.5 h of incubation at 28°C.
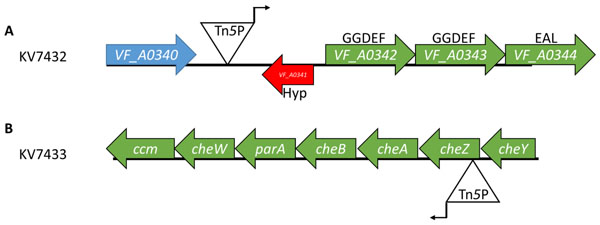
The location and orientation of the Tn5P insertions in two motility mutants. The insertion in KV7432 is located in the intergenic region between VF_A0340 and VF_A0341 with the A1/34 promoter oriented toward VF_A0341 (A). The insertion in KV7433 is located within the 5’ end of cheZ with the A1/34 promoter oriented toward cheA (B).
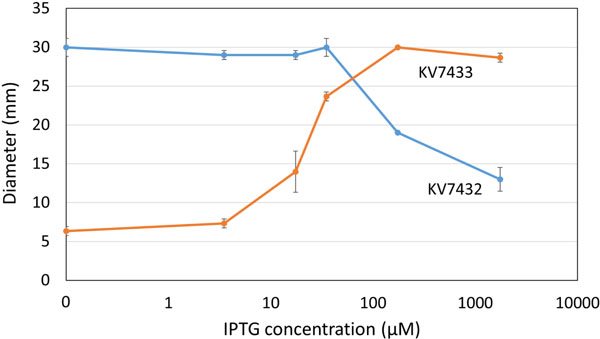
Dependence of mutant motility phenotypes on IPTG. Motility mutants KV7432 and KV7433 were grown at 28°C in TB-SW motility medium containing the indicated concentrations of IPTG. The average diameter of migration of triplicate samples after 5 hours is shown. Standard deviation is indicated by error bars. Error bars smaller than the plotted points are occluded by the points and not visible.
Plasmid pJMO10, which was used to deliver the Tn5+ promoter (Tn5P) for transposon mutagenesis, was constructed as follows. Oligonucleotides 1439 and 1440, which contained sequences for the LacI-repressible PA1/34 promoter [1, 30], were annealed and ligated into the ApaI/SpeI-digested Tn5 delivery plasmid pEVS170 [31]. The insertion of the promoter into Tn5 was confirmed by sequencing.
Wrinkled colony formation. Strains were grown overnight in LBS containing CM at 28oC with agitation, then subcultured into fresh medium and grown until they reached exponential phase. Cultures were then diluted to an optical density at 600 nm (OD600) of 0.2, concentrated by centrifugation, and re-suspended in fresh medium. 10 µl aliquots of each culture were spotted onto an agar plate and incubated at room temperature. Images of the wrinkled spots were captured at the indicated times.
Tn5P mutagenesis and identification of IPTG-inducible motility mutants. Parental strain KV6576 was mutagenized with Tn5P by performing a tri-parental conjugation [32] with two E. coli strains, one carrying pJMO10 and other carrying pEVS104 [32] and selecting for the insertion of the transposon using Erm-containing LBS (LBS-erm) plates. Resulting colonies were inoculated into TB-SW motility plates lacking or containing 1.75 mM IPTG. Putative motility mutants with IPTG-controlled phenotypes were purified from motility plates by streaking onto LBS-erm plates. These strains were then evaluated a second time to verify their motility phenotypes.
Identification of Tn5P insertion sites. To determine the site of the Tn5P insertion in the motility mutants, chromosomal DNA was isolated and digested using the HhaI restriction enzyme as previously described [31]. Digested DNA was then self-ligated and transformed into CC118 cells, and clones were selected on Erm-containing BHI agar. The resulting plasmids were sequenced using primer 908.
Motility Assays. Motility mutants were grown overnight in LBS and the cultures were diluted to an OD600 of 0.4. Cells were pelleted, washed, and re-suspended in LBS and 5 µl aliquots were inoculated on the surface of TB-SW motility plates that contained the indicated amounts of IPTG. Representative images were captured and the diameters of the outer ring of the swimming cells were measured using a ruler.
RESULTS AND DISCUSSION
Generation of Tn5P, a Tn5 transposon with an outward-facing promoter. One genetic tool that remained to be developed for V. fischeri was a method for randomly inserting an inducible promoter into the genome. To develop this tool, we chose to use the LacI-repressible promoter A1/34 [1]. This promoter contains two LacI binding sites, one between the -35 and -10 sites and another that overlaps the transcriptional start site (Fig. 1), and has been shown previously to function as a strong promoter in V. fischeri when it was inserted directly upstream of an operon [1]. We then engineered the mini-Tn5 delivery vector pEVS170 [31] to contain this promoter within the transposable element in an outward-facing position; the insertion of the promoter was confirmed by sequencing. This transposon will be referred to as Tn5P, for Tn5 plus Promoter.
Insertion of the lacI gene into V. fischeri and phenotypic assessment. V. fischeri does not contain the lac operon or the lacI repressor gene. Therefore, to control expression from the promoter within Tn5P, it was necessary to introduce the lacI gene into V. fischeri. One location traditionally used to insert genes within the V. fischeri genome is the Tn7 site, which is positioned between yeiR and glmS (Fig. 2) [15, 33]. However, because this site is heavily utilized for single copy complementation (e.g., [34]), we chose to leave this site intact for future manipulations. Instead, we targeted the insertion of lacIq, a more highly transcribed allele of the lacI gene, to a region immediately adjacent to the Tn7 site (between yeiR and the Tn7 site) (Fig. 2). The result was a strain, KV6576, that contains lacIq, retains an intact Tn7 site, and remains unmarked, allowing for the use of an antibiotic resistance marker in future manipulations.
With the generation of the lacIq-containing V. fischeri, two questions arose: (i) Is the lacIq allele functional (e.g., does it control gene expression in V. fischeri?); and (ii) is the Tn7 site, which is adjacent to the site of insertion of the lacIqgene, still permissive for insertion events? To assess whether the lacIq allele in KV6576 was functional, we investigated the ability of this strain to impact expression of a lac promoter-controlled gene. Specifically, we introduced into KV6576 a plasmid, pCLD46, that contains the rscS gene driven by the lac promoter, or pVSV105, the empty vector from which pCLD46 was derived. When pCLD46 is introduced into wild-type V. fischeri, RscS protein is made and induces biofilm formation [13]. One biofilm phenotype that can be readily assessed is the formation of wrinkled colonies. We anticipated that, if the lacIq allele in KV6576 were functional, then LacI would repress expression of rscS, resulting in a strain that either fails to form wrinkled colonies or does so after a delay. Indeed, we found that this strain failed to wrinkle after 24 hours of growth in the absence of IPTG (Fig. 3). Moreover, when we grew the strain in the presence of IPTG, which should inactivate LacI, wrinkled colonies developed with a timing indistinguishable from the control (Fig. 3 and data not shown). These data indicate that functional LacI was made and was responsive to IPTG. We note, however, that the repression of rscS expression from pCLD46 was not complete: at later times, the strain exhibited a modest wrinkling phenotype in the absence of IPTG (Fig. 3). We attribute this result to the inability of LacI expressed from the chromosome to fully repress a promoter present on a multi-copy plasmid.
To verify that the Tn7 site near the site of lacIq insertion in KV6576 remained amenable to manipulation, we used pEVS107, a Tn7 delivery vector that targets the Tn7 site [15], to introduce an Erm resistance cassette at that location. pEVS107 contains an Erm resistance cassette within the Tn7 ends and a kanamycin resistance cassette outside. Erythromycin-resistant strains were readily isolated and exhibited sensitivity to kanamycin, as expected when the Tn7 cassette inserts at the Tn7 site (data not shown).
Identification of motility mutants. Our data above indicate that the lacI gene inserted into the chromosome is functional to repress transcription of a lac promoter-controlled gene. However, the question remained, does the lacIq allele control transcription from the PA1/34 promoter contained within Tn5P? Specifically, we wondered whether we could induce or repress native V. fischeri genes in KV6576 containing insertions of Tn5P. To address this question, we chose to evaluate a readily assessable phenotype, motility. V. fischeri contains a number of genes known [35-38] or predicted [39] to impact motility. We hypothesized that Tn5P insertions upstream of such genes could result in strains with inducible or repressible motility. We thus introduced Tn5P into KV6576 and assessed mutant motility on soft agar plates that contained or lacked IPTG. From a screen of about 2000 mutants, we identified about 20 strains with potential IPTG-dependent motility phenotypes and confirmed the phenotypes of a subset of these mutants. Of these, our focus was drawn to two strains with opposing phenotypes (Fig. 4). One strain, KV7432, had IPTG-repressible motility: it exhibited near wild-type motility in the absence of IPTG but greatly diminished motility in the presence of IPTG (Fig. 4B). In contrast, IPTG did not impact motility of the control strain (Fig. 4A). The second strain of interest, KV7433, had IPTG-inducible motility: it was non-motile in the absence of IPTG but regained a wild-type motility phenotype in the presence of IPTG (Fig. 4C). Because of their strong yet opposite phenotypes, we chose these two strains for additional characterization.
We first identified the sites of insertion of Tn5P as described in Materials and Methods. The mutant with IPTG-repressible motility, KV7432, contained an insert within the intergenic region between VF_A0340 and VF_A0341, with the promoter of Tn5P oriented toward VF_A0341 (Fig. 5A). In this orientation, the promoter appears positioned to drive expression of the nearby three-gene operon consisting of VF_A0342, VF_A0343, and VF_A0344. These three genes are predicted to encode proteins with GGDEF or EAL domains. These domains are found in diguanylate cyclase and phosphodiesterase proteins, which synthesize and degrade, respectively, the second messenger cyclic-di-GMP (c-di-GMP) [40]. High levels of cellular c-di-GMP inhibit motility in a variety of bacteria [41]. Therefore, we hypothesize that IPTG-mediated expression from the transposon promoter increases the levels of c-di-GMP in the cell and thus, inhibits motility. It also remains possible that expression from the Tn5P promoter decreases the expression of VF_A0341, which encodes a hypothetical protein with no conserved domains.
The mutant with IPTG-inducible motility, KV7433, carried the Tn5P insertion within the cheZ gene, with the transposon’s promoter oriented with the che operon (Fig. 5B). In the absence of IPTG, the transposon insertion should interrupt transcription of cheZ as well as the downstream che genes which coordinate chemotaxis and are required for motility in V. fischeri [38]. Thus, it was not surprising that, in the absence of IPTG, this mutant exhibited a motility defect. Given that the Tn insertion was within the cheZ gene, however, it was unexpected that the addition of IPTG would restore near wild-type motility (Fig. 4). Upon closer investigation of the insertion site, we noted that (1) the Tn is inserted near the beginning of cheZ, and (2) an ATG start codon within the Tn5P transposon end is in frame with the cheZ open reading frame (Fig. 1B and data not shown). Based on these observations, we hypothesize that expression from the transposon’s promoter and translation from the ATG within the transposon end results in the production of a hybrid CheZ protein with an altered N-terminus that is functional to promote motility.
Assessment of IPTG Induction. Our previous experiments used a single concentration of IPTG, 1.75 mM, to induce transcription from the Tn5P promoter. However, it was unclear whether this high amount of IPTG was necessary to obtain full repression/induction of motility by our strains. Thus, to determine the sensitivity of the Tn5P promoter to IPTG, we assessed the mutants’ motility phenotypes in the presence of a range of IPTG concentrations. KV7433 exhibited a dose-dependent increase in motility within a wide range of IPTG concentrations between 3.5 µM and 175 µM IPTG that was not further increased with additional IPTG (Fig. 6). The other mutant, KV7432, similarly exhibited a dose-dependent change. In this case, the impact on motility required higher IPTG concentrations, above 35 µM; at the highest amount tested, 1.75 mM, motility was not fully repressed (Fig. 6). These data further support our conclusion that expression from the Tn5P promoter is induced by the addition of IPTG. Additionally, because we obtained different ranges of IPTG addition required for a transition from the motile to non-motile phenotype in the two strains, we conclude that it may be necessary to experimentally determine the optimal expression of a particular gene obtained using this experimental set-up (lacIq/Tn5P) by titrating the concentration of IPTG in the medium against the phenotype being tested.
CONCLUDING REMARKS
We have developed a means to randomly insert and control an inducible promoter within the chromosome of V. fischeri by (1) generating a mini-Tn5 modified to contain an outward-facing, LacI-repressible promoter and (2) engineering a background strain that expresses LacI to control the promoter. Insertion of the Tn5P within a gene or locus will result in disruption of the gene or locus and has the additional ability to conditionally induce transcription of downstream genes, thus reducing the problem of polarity typically associated with Tn insertions. Finally, our evidence indicates that the Tn5P end may, in some cases, generate and express proteins with N-terminal truncations. Thus, this genetic tool increases the number of phenotypes V. fischeri researchers can screen and has the potential to greatly increase our understanding of the physiology of this model organism.
CONFLICT OF INTEREST
This work was supported by Loyola University Chicago Research funding council and to NIH grant 2R01 GM059690 awarded to KLV.
ACKNOWLEDGEMENTS
We thank Eric Stabb for suggesting the use of the A1/34 promoter for this study, and members of the Visick lab for critically reading this manuscript.

