All published articles of this journal are available on ScienceDirect.

Microbial Community Structure Across a Wastewater-Impacted Riparian Buffer Zone in the Southeastern Coastal Plain
Abstract
Riparian buffer zones are important for both natural and developed ecosystems throughout the world because of their ability to retain nutrients, prevent soil erosion, protect aquatic environments from excessive sedimentation, and filter pollutants. Despite their importance, the microbial community structures of riparian buffer zones remains poorly defined. Our objectives for this study were twofold: first, to characterize the microbial populations found in riparian buffer zone soils; and second, to determine if microbial community structure could be linked to denitrification enzyme activity (DEA). To achieve these objectives, we investigated the microbial populations of a riparian buffer zone located downslope of a pasture irrigated with swine lagoon effluent, utilizing DNA sequencing of the 16S rDNA, DEA, and quantitative PCR (qPCR) of the denitrification genes nirK, nirS, and nosZ. Clone libraries of the 16S rDNA gene were generated from each of twelve sites across the riparian buffer with a total of 986 partial sequences grouped into 654 operational taxonomic units (OTUs). The Proteobacteria were the dominant group (49.8% of all OTUs), with the Acidobacteria also well represented (19.57% of all OTUs). Analysis of qPCR results identified spatial relationships between soil series, site location, and gene abundance, which could be used to infer both incomplete and total DEA rates.
INTRODUCTION
Riparian buffers are a best management practice utilized extensively in the protection of freshwater ecosystems from agriculturally-generated nitrate (NO3-) pollution [1, 2]. While there are several routes for NO3- removal from riparian buffer zones, denitrification is thought to serve as the primary mechanism [3, 4]. Denitrification is the biological reduction of NO3- or nitrite (NO2-) under anoxic conditions to either nitrous oxide (N2O) or dinitrogen (N2) gases. This process is achieved through the following series of enzymatic steps: [5]; the resultant gases are then permanently removed from the ecosystem. However, despite this vast knowledge of the relationship between riparian buffers and denitrification, there is a paucity of information in regards to riparian buffer microbial community composition. Equally lacking is an understanding of the relationships between these microbial communities and the biological processes – such as denitrification – that occur within the riparian buffer.
Earlier studies suggested a relationship between microbial community composition and denitrification in agricultural soils [6, 7]. Likewise, Martin et al. and Rich and Myrold examined microbial community composition and denitrification in riparian buffer soils [8, 9]. However, none of these studies were performed in riparian buffers adjacent to areas heavily NO3- loaded. Riparian buffers located next to fields receiving high NO3- loads are of particular importance because riparian buffers have a higher propensity for nitrous oxide (N2O) emissions [10-12]. Emissions of N2O, a potent greenhouse gas, play a critical role in the climate change discussion due to its ability to degrade stratospheric ozone [13]. Therefore, in order to better understand denitrification and N2O emissions in riparian soils receiving high loads of nitrate, it is important to examine the microbial community composition of these soils. Such studies would provide insight into whether microbial community composition exerts direct control on the denitrification process and emission of N2O.
While denitrifiers make up a sizeable portion of soil microbial communities (up to 5%) [5], culturing techniques are rarely able to identify greater than 1% of an environments microbial content [14]. To counter the problems with culturing methods, 16S rDNA gene sequencing is routinely used to provide descriptive overviews of complex microbial communities, something that is currently lacking in the analysis of riparian buffers. Likewise, the use of quantitative Real-Time PCR (qPCR) using primers specific for genes involved in microbial processes allows for the quantification of specific groups of microbial organisms in a variety of ecosystems without the need for culturing.
In this study we examined the microbial communities of a riparian buffer located adjacent to a heavily loaded swine wastewater spray field. Our objectives for this study were to: 1) characterize the microbial populations found in the soils of this riparian buffer zone; and, 2) determine if differences in microbial community structure could be linked to denitrification enzyme activity (DEA) rates.
MATERIALS AND METHODS
Site Description
The riparian zone studied was located on an unnamed tributary within the Herrings Marsh Run (HMR) watershed. The HMR is a 23.6 km2 watershed located in the coastal plain region of Duplin County, North Carolina. The taxonomic classes of the soils varied throughout the riparian zone and are typical of those found of the Coastal Plain (Table 1). The riparian zone had a variable width of 5 to 30 meters and a stream span of 200 meters. The riparian landscape was split into two portions: 1) a restored riparian zone (see RRZ, Fig. 1) located closest to the swine wastewater spray field; and 2) a natural riparian zone (see NRZ, Fig. 1) containing native vegetation located on the opposite side of the stream [12]. The site is more fully described in Hunt et al. (2004). The restored riparian landscape was used to minimize the effect of surface and ground water outflows from a coastal bermudagrass (Cynodon dactylon (L.) Pers.) pasture irrigated with swine lagoon effluent which was located upslope (see SWSF, Fig. 1).
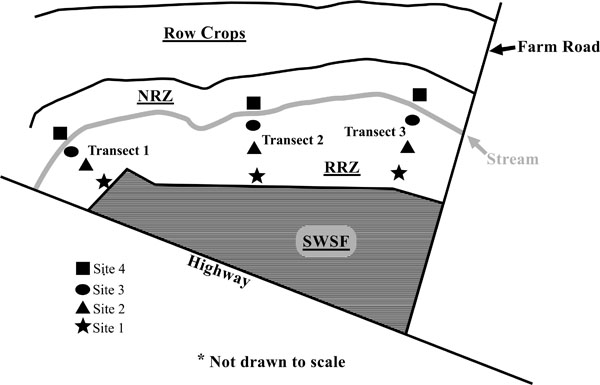
A schematic of the experimental site including swine wastewater spray field (SWSF), restored riparian zone (RRZ), natural riparian zone (NRZ), stream, and soil sampling sites.
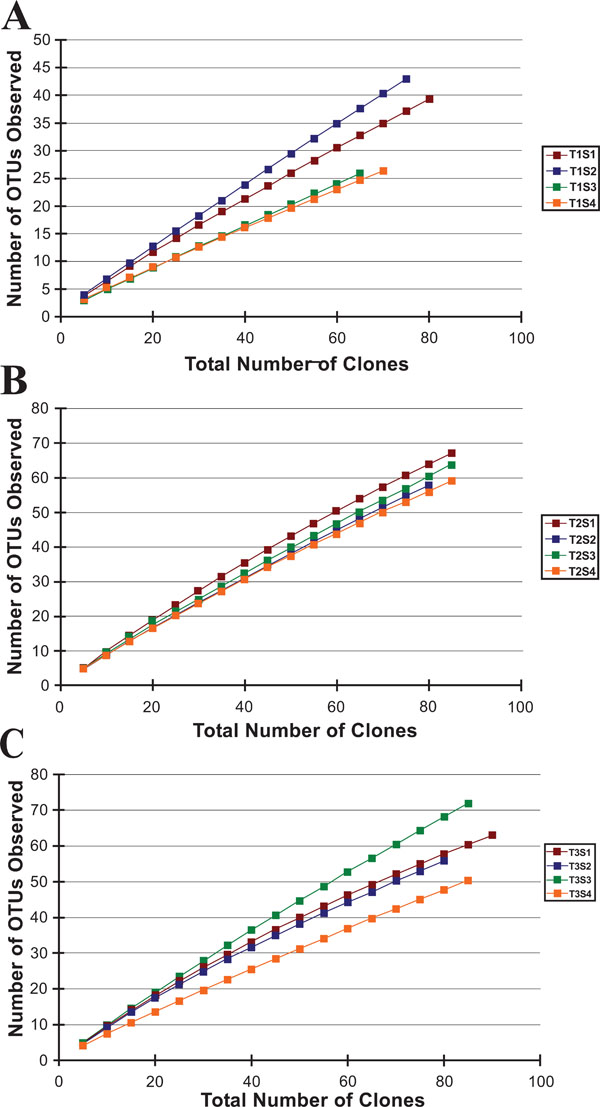
Rarefaction curves from the three riparian buffer zone transects analyzed in this study.
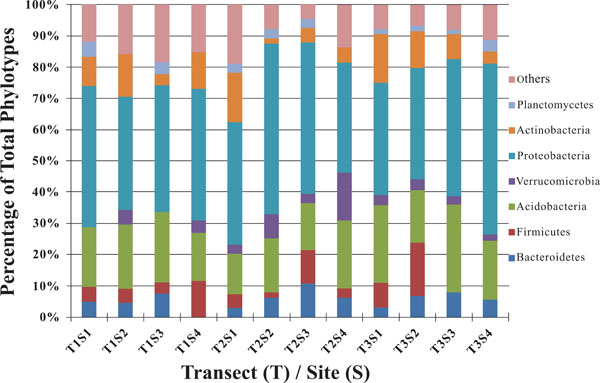
Taxonomic breakdown of classified 16S rDNA gene sequences from riparian buffer zone.
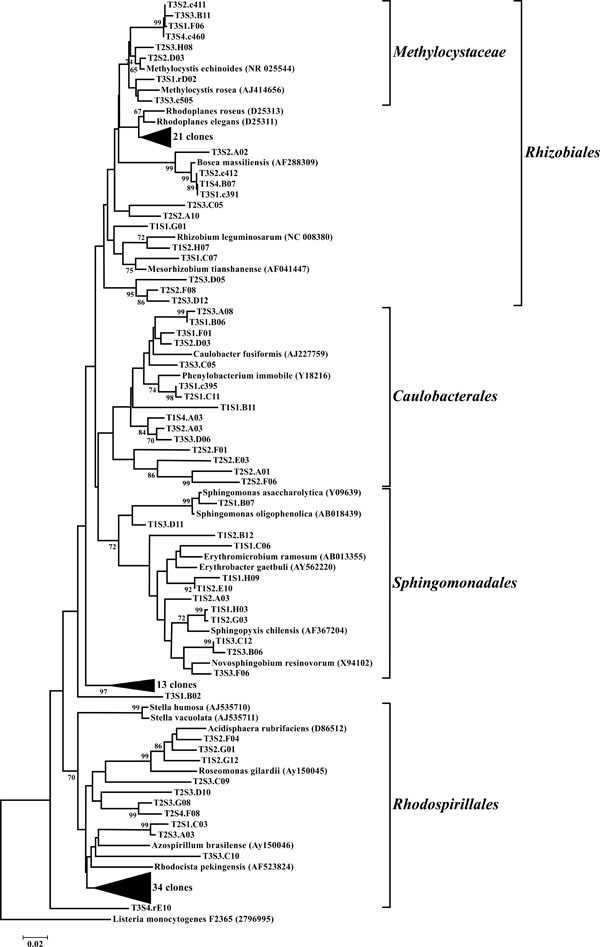
Neighbor-joining tree of α-Proteobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
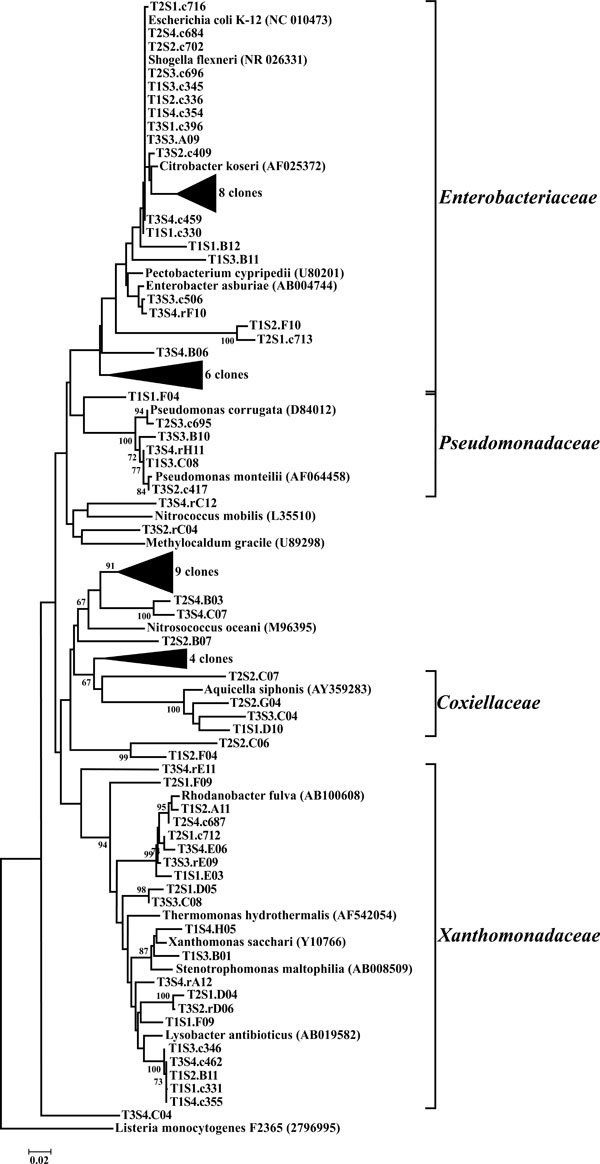
Neighbor-joining tree of γ-Proteobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
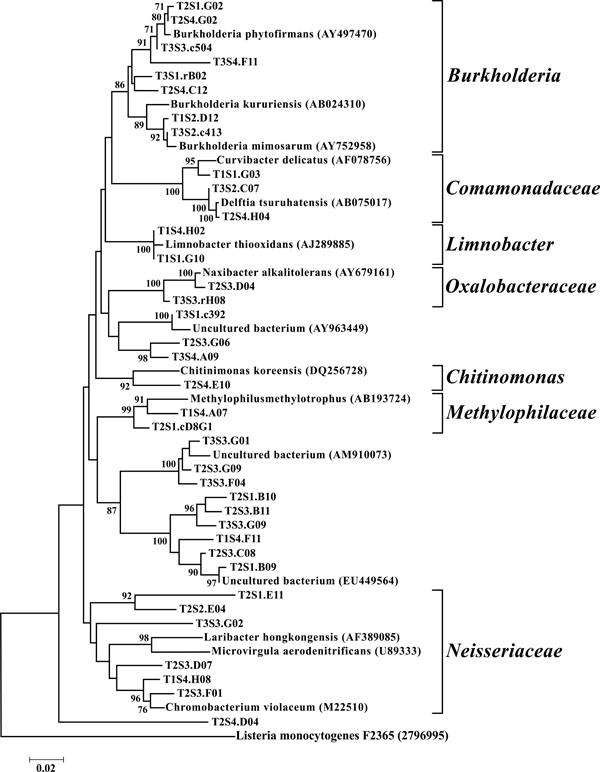
Neighbor-joining tree of β-Proteobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
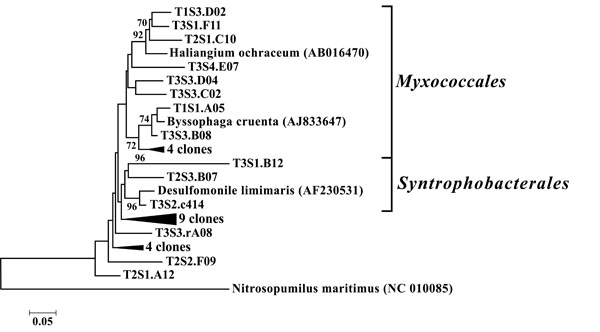
Neighbor-joining tree of δ-Proteobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
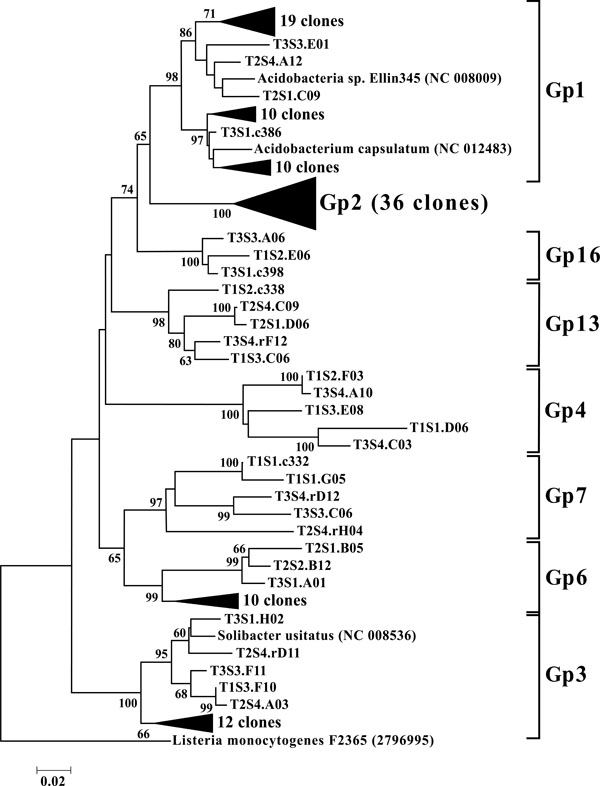
Neighbor-joining tree of Acidobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
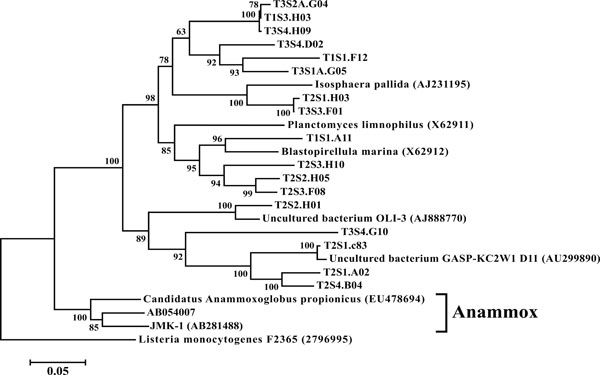
Neighbor-joining tree of Planctomycetes. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
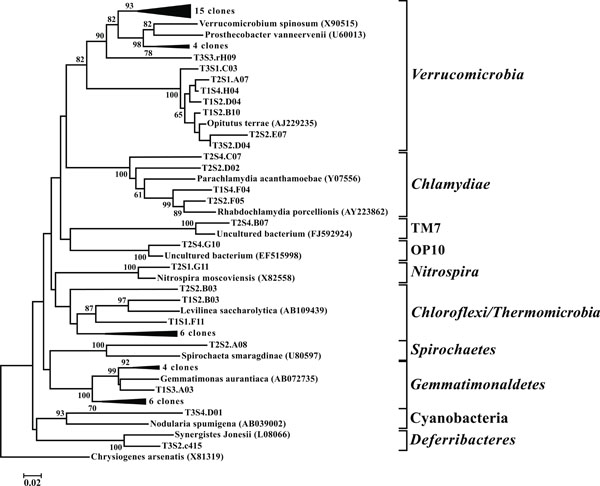
Neighbor-joining tree of other taxonomic groups. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
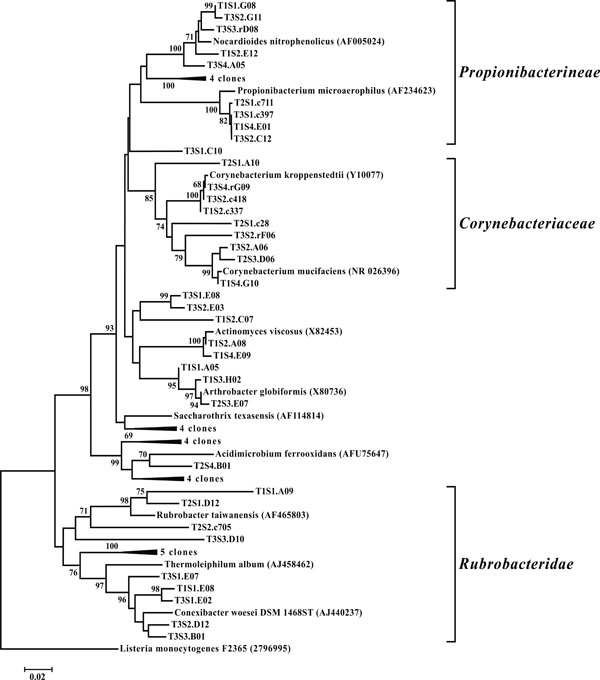
Neighbor-joining tree of Actinobacteria. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
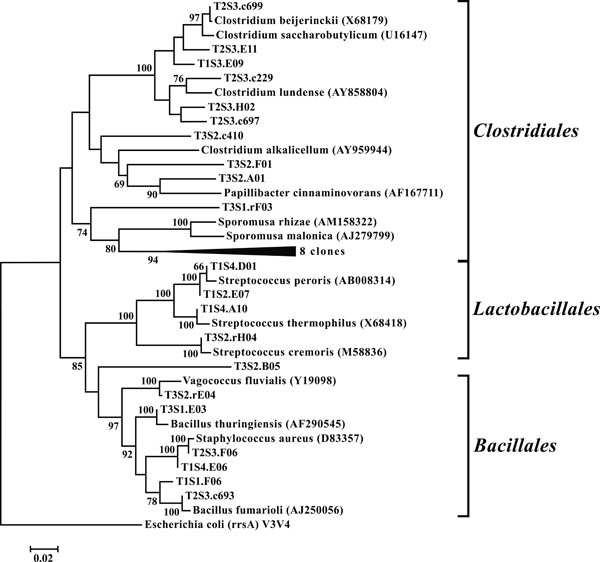
Neighbor-joining tree of Firmicutes. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
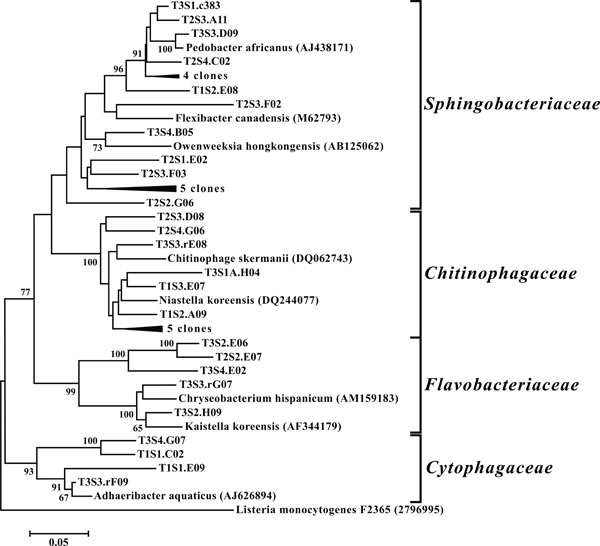
Neighbor-joining tree of Bacteroidetes. The frequency (%) of which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of the bootstrap replicates.
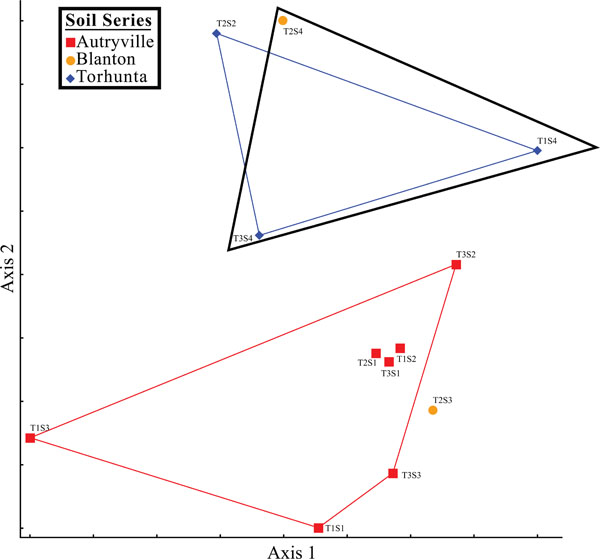
Nonmetric multidimensional scaling graph.
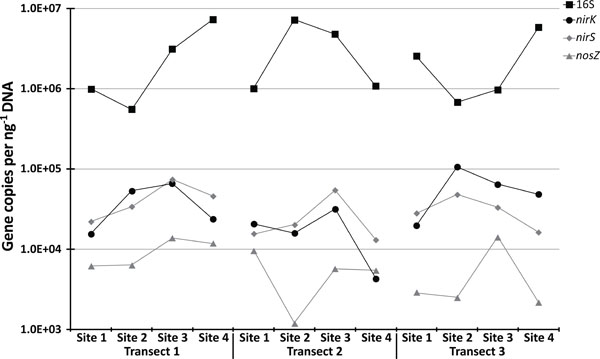
qPCR results.
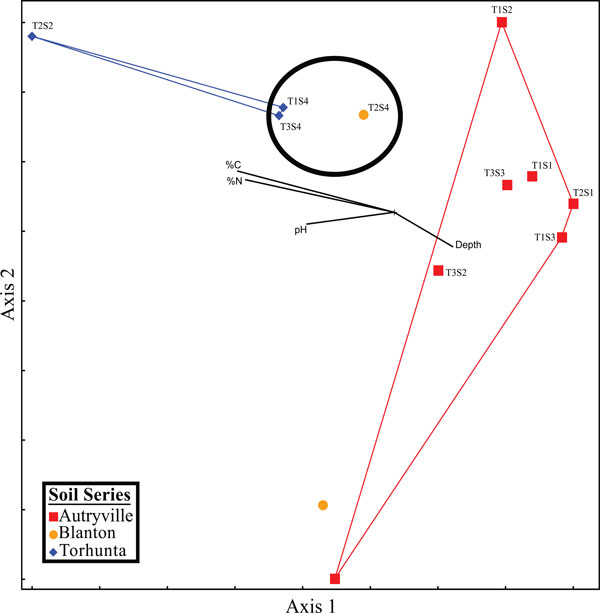
NMS graph of qPCR.
Soil Characteristics
| Transect (T) Site (S) | Taxonomic Class† | Water Table Depth‡ | DEA Incomplete | DEA Total | Carbon (%) | Nitrogen (%) | C:N Ratio | pH | EC (µS/cm) |
|---|---|---|---|---|---|---|---|---|---|
| ng N2O-N/gm Soil/hr | |||||||||
| T1S1 | A | 137 | 25.1 ± 17.3 | 37.6 ± 46.1 | 2.41 | 0.11 | 21.9 | 3.69 | 64.9 |
| T1S2 | A | 75 | 0 ± 0 | 5.0 ± 8.1 | 3.79 | 0.17 | 22.2 | 3.85 | 57.5 |
| T1S3 | A | 75 | 22.8 ± 7.3 | 117.7 ± 22.7 | 5.79 | 0.26 | 22.2 | 3.99 | 40.1 |
| T1S4 | C | 45 | 144.8 ± 103.1 | 289.1 ± 299.5 | 14.78 | 0.74 | 20.0 | 4.75 | 73.4 |
| T2S1 | A | 167 | 2.5 ± 3.0 | 23.1 ± 16.0 | 1.11 | 0.064 | 17.3 | 5.35 | 34.1 |
| T2S2 | C | 45 | 172.5 ± 175.4 | 610.4 ± 389.0 | 26.53 | 1.31 | 20.2 | 5.67 | 82.2 |
| T2S3 | B | 45 | 24.6 ± 28.6 | 129.0 ± 152.7 | 8.64 | 0.51 | 16.9 | 5.52 | 71.8 |
| T2S4 | B | 90 | 73.6 ± 19.1 | 153.7 ± 42.5 | 4.62 | 0.21 | 22.3 | 4.17 | 143.1 |
| T3S1 | A | 168 | 0 ± 0 | 5.9 ± 1.0 | 1.09 | 0.065 | 16.8 | 4.56 | 20.9 |
| T3S2 | A | 75 | 0 ± 0 | 0.9 ± 0.8 | 1.67 | 0.090 | 18.6 | 4.05 | 43.5 |
| T3S3 | A | 75 | 0 ± 0 | 5.9 ± 0.8 | 1.71 | 0.083 | 20.6 | 4.06 | 63.3 |
| T3S4 | C | 75 | 137.6 ± 58.9 | 488.7 ± 131.2 | 30.88 | 1.71 | 18.0 | 4.23 | 220 |
† A = Autryville, loamy, siliceous, subactive, thermic Arenic Paleudults, 0–6% slope
B = Blanton, loamy, siliceous, semiactive, thermic Grossarenic Paleudults, 0-6% slope
C = Torhunta, coarse-loamy, siliceous, active, acid, thermic Typic Humaquepts, 0-2% slope
‡ centimeters below surface
Primers and Plasmids Used in this Study
| Primers | Sequence (5’ to 3’) | Target | Tm† | Reaction Tm | Reference |
|---|---|---|---|---|---|
| amoA-1F | GGGGTTTCTACTGGTGGT | amoA | 54.1 °C | 54 °C | [47] |
| amoAr NEW | CCCCTCBGSAAAVCCTTCTTC | 58.8 °C | [48] | ||
| 1F_nirK | GGMATGGTKCCSTGGCA | nirK | 58.0 °C | 53 °C | [49] |
| nirK5R | GCCTCGATCAGRTTRTGG | 52.8 °C | [49] | ||
| cd3aF_nirS | GTSAACGTSAAGGARACSGG | nirS | 57.1 °C | 55 °C | [50] |
| R3cd_nirS | GASTTCGGRTGSGTCTTGA | 55.8 °C | [50] | ||
| nosZF | CGYTGTTCMTCGACAGCCAG | nosZ | 58.6 °C | 55 °C | [51] |
| nosZ-1622R | CGSACCTTSTTGCCSTYGCG | 63.1 °C | [50] | ||
| 515F | TGCCAGCAGCCGCGGTAA | 16S v4-v5 region | 63.3 °C | 55 °C | [52] |
| 927R | CTTGTGCGGGCCCCCGTCAATTC | 65.1 °C | [53] | ||
| Accession No. | |||||
| Plasmids | Characteristics | ||||
| pCPDamoA1 | pCR4.1-TOPO carrying amoA fragment | HQ674785 | [25] | ||
| pCPDnirS1 | pCR4.1-TOPO carrying nirS fragment | HQ674783 | [25] | ||
| pCPDnirK1 | pCR4.1-TOPO carrying nirK fragment | HQ674782 | [25] | ||
| pCPDnosZ1 | pCR4.1-TOPO carrying nosZ fragment | HQ674784 | [25] | ||
| pCPDv4v5 | pCR4.1-TOPO carrying 16S fragment | HQ674781 | [25] |
† Tm = Melting temperature
Results of 16S rDNA Gene Libraries
| Site | Library | Number of Sequences (N) | OTUsa (O) |
|---|---|---|---|
| Transect 1 Site 1 | T1S1 | 84 | 42 |
| Transect 1 Site 2 | T1S2 | 77 | 44 |
| Transect 1 Site 3 | T1S3 | 68 | 27 |
| Transect 1 Site 4 | T1S4 | 72 | 26 |
| Transect 2 Site 1 | T2S1 | 85 | 69 |
| Transect 2 Site 2 | T2S2 | 76 | 64 |
| Transect 2 Site 3 | T2S3 | 77 | 66 |
| Transect 2 Site 4 | T2S4 | 90 | 65 |
| Transect 3 Site 1 | T3S1 | 91 | 64 |
| Transect 3 Site 2 | T3S2 | 85 | 59 |
| Transect 3 Site 3 | T3S3 | 91 | 75 |
| Transect 3 Site 4 | T3S4 | 90 | 53 |
a – Based on 97% sequence similarity
Soil Sample Collection and Analysis
Soil samples were collected from four sites on three transects (three sites in the restored riparian zone and one site in the natural riparian zone) in October 2006. Soil surface samples (5 cm diameter, 15 cm depth) were collected in triplicate. Samples were placed in plastic bags, stored on ice, transported to the laboratory, and stored overnight at 4°C. Total carbon and nitrogen were determined by analysis with a Model CN2000 analyzer (LECO, St. Joseph, MI). Soil pH and EC values were determined using a 1:2 ratio of soil to deionized water and measured with an AP50 pH/Ion/Conductivity meter (Denver Instrument Company, Arvada, CO).
Denitrification Enzyme Activity
Denitrification enzyme activity (DEA) was measured by the acetylene blockage method [5] as modified by Hunt [12]. All analyses were performed in triplicate runs, with each run containing two bottles per sample. Bottles measuring incomplete denitrification, received 5 mL of chloramphenicol (1 g L-1) to inhibit protein synthesis, whereas bottles measuring total denitrification, received 5 mL of chloramphenicol (1 g L-1) as well as 15 mL acetylene (produced from calcium carbide) to block denitrification at the nitrous oxide phase addition. Rates of N2O accumulation were expressed on a dry soil weight basis.
DNA Extraction
DNA from each site was extracted from 1.0 g of soil using a SoilMaster DNA Extraction Kit (Epicentre, Madison, WI) according to manufacturer specifications. This extraction was further treated to remove the PCR-inhibiting, humic acid contaminants. To this end, the entire SoilMaster prep was concentrated to a total volume of 50 µl, mixed with 6 µl DNA loading buffer (20% w/v sucrose, 0.01% w/v Orange G), and run on a 0.5% agarose gel for 45 minutes. The gel was stained with SYBR Safe to visualize the DNA, excised from the gel, and purified using an UltraClean GelSpin DNA Extraction Kit (MO BIO Laboratories, Inc., Carlsbad, CA) according to manufacturer specifications. Final DNA quantity and quality were determined via Biophotometer (Eppendorf, Hamburg, Germany), and electrophoresis on a 1% agarose gel stained with SYBR Safe (Invitrogen, Carlsbad, CA).
16S rDNA Gene Library Construction, Sequence, and Phylogenetic Analysis
Primers used for 16S rDNA gene library construction are listed in Table 2. DNA sequences were analyzed and edited using Geneious (version 4.5.4, Biomatters Ltd., Auckland, New Zealand, [15]). Chimeric sequences were identified using the programs CHECK_CHIMERA [16] and Bellerophon [17]. Sequence alignments were performed using the MUSCLE plugin of Geneious [18], and manually verified. The 16S rDNA gene sequences were compared to sequences in the GenBank database by using BLAST (Basic Local Alignment Sequence Tool) [19]. To determine their approximate taxonomical classifications, 16S rDNA gene sequences were analyzed at the Ribosomal Database Project using the Naïve Bayesian Classifier [20]. Rarefaction curves were calculated using Analytic Rarefaction (version 1.3, [21]). Prior to regression analyses relative abundances were arcsine transformed to reduce skew [22]. Phylogenetic reconstructions were performed in MEGA (version 4.0, [23]) using the neighbor-joining (NJ) algorithm, with bootstrap values calculated from 1,000 replicate runs. Evolutionary distances were computed using the Tajima-Nei method, and positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons. For all figures of phylogenetic reconstructions, the frequency (%) with which a given branch was recovered in 1,000 bootstrap replications is shown above branches recovered in more than 65% of bootstrap replicates.
Quantitative real-time PCR (qPCR) Assays
All qPCR assays were performed using the LightCycler 480 Real-Time PCR Detection System (Roche Diagnostics, Indianapolis, IN). Abundances were measured using primers synthesized by Integrated DNA Technologies (Coralville, IA). Fragments of 16S, nirS, nirK and nosZ were amplified via qPCR using the primer pairs listed in Table 2. Assays were carried out using SYBR GreenER qPCR SuperMix (Invitrogen, Carlsbad, CA) in a total volume of 25 µL. The final reaction concentration of reagents was as follows: 1X SYBR GreenER qPCR SuperMix; 200 nM each of forward and reverse primers; and 10 ng of DNA template. The qPCR reaction conditions were as follows: (1) an initial denaturation at 95°C for 5 min; (2) 50 cycles of denaturation at 95°C for 30 sec, an annealing temperature as indicated in Table 2 for 30 sec, and elongation at 72°C for 30 sec; (3) melting curve analysis to confirm amplification product specificity. Fluorescent measurements were taken during the annealing phase of each cycle. Data was collected and processed using the LightCycler 480 software package. All qPCR assays included control reactions without template. Each assay also contained appropriate standard DNA reactions with concentrations between 101 and 109 copies per reaction, and were used to calculate amplification efficiencies according to the equation: E = 10[-1/slope] [24]. DNA standards consisted of plasmids carrying the appropriate target gene that were previously sequenced to confirm their identity and primer binding sites [25]. Each assay was performed in triplicate, with duplicate measurements for each sample.
Statistics
Prior to statistical analysis, all gene abundances were normalized to the amount of DNA collected per sample and log10 transformed. Corrections for copies per organisms were also factored into the final gene abundance rates, with 1 copy per organism for the denitrification genes nirK, nirS, and nosZ [26], and 3.6 copies per organism for the 16S rDNA gene [27]. Linear regressions and other statistical analyses were performed using SAS version 9.2 (SAS Inst., Cary, NC). Nonmetric multidimensional scaling (NMS) analyses were performed using PCORD version 6 (MJM Software, Gleneden Beach, OR). Use of phylogenetic data for NMS analyses were based on presence/absence, as opposed to relative abundance, of each genus.
Nucleotide Sequence Accession Numbers
Nucleotide sequences determined in this study have been deposited in the GenBank database under accession numbers KC786278 - KC786879.
RESULTS AND DISCUSSION
Physicochemical Characteristics of the Riparian Buffer Zone
Characteristics of the riparian buffer zone are displayed in Table 1. Soil taxonomy was typical of coastal plain soils, with three soil series classified: Autryville; Blanton; and Torhunta. Depth to the water table ranged from 168 centimeters at the highest point of the riparian buffer zone (T3S1) to 45 centimeters at the lowest points (T1S4, T2S2, and T2S3). Carbon and nitrogen ranged from 1.09 to 30.88% and 0.065 to 1.71% of total soil weight respectively, and carbon to nitrogen (C:N) ratios were consistent, with a range of 16.9 to 22.3. The soils of all three transects were acidic, ranging from 3.69 to 5.67. Electrical conductivity ranged from 20.9 to 220 µS/cm.DEA was variable across the riparian buffer: incomplete DEA ranged from 0 to 172.5 ng N2O-N gm soil h-1; total DEA ranged from 0.9 to 610.4 N2O-N gm soil h-1. Previously Hunt et al. demonstrated that carbon, depth to water table, and nitrogen were the most highly correlated factors to DEA [12]. Regression analysis of DEA against the riparian buffer zone physicochemical characteristics revealed that carbon was the most important factor for both incomplete (r2 = 0.799; P < 0.0001) and total (r2 = 0.930; P < 0.0001) DEA.
Construction of 16S rDNA gene Libraries
Soil microbial communities from each of the twelve sites were analyzed by PCR amplification of total DNA extracts with universal bacterial primers. For each of the twelve libraries obtained, a total of 96 clones were selected randomly for sequencing of the V4 and V5 regions of the 16S rDNA gene. A total of 1,152 clones were sequenced, 27 of which were omitted from the study after being identified as putative chimeras by CHIMERA_CHECK and Bellopheron, with an additional 17 removed as sequencing failures. Removal of redundant sequences (100% identity) from the same site only, identified a total of 986 novel, partial 16S rDNA sequences for use in downstream analysis. Comparison of clone sequences with GenBank using BLAST revealed sequence similarities between the ranges of 88 and 100 percent.
Bacterial Community Analysis
After processing, the partial 16S rDNA gene sequences were binned into operational taxonomic units (OTUs) based on a >97% sequence similarity to other sequences from the same sample site; this resulted in a total of 654 OTUs (Table 3). We opted for a >97% sequence similarity threshold for two reasons: first, it generally correlates with the 70% DNA-DNA hybridization binding criterion used as the gold standard to identify new bacterial species [28]; and second, it is a frequently used taxonomic threshold, allowing for comparison to other data sets. After generation of the OTUs, the bacterial libraries were analyzed down to the genus level. For all libraries the rarefaction curves failed to plateau, indicating that within these samples the number of sequenced clones failed to exhaust the microbial diversity (Fig.2). Phylogenetic comparison was also performed on all classifiable OTUs to closely related isolates (Fig. 4through 13).
(i) Proteobacteria
The dominant bacterial division represented in the libraries was the Proteobacteria that accounted for 42.8% (280 of 654) of all OTUs (Fig. 3). When the Proteobacteria were categorized by subclass, the α- and γ- subclasses were the major constituents, accounting for 19.42% (127 OTUs; Fig. 4) and 13.00% (85 OTUs; Fig. 5) of all OTUs respectively. The β- (37 OTUs, 5.66%; Fig. 6) and δ- (24 OTUs, 3.67%; Fig. 7) proteobacterial OTUs were represented in lesser proportions. The identification of a large number of OTUs classified as α-Proteobacteria has been demonstrated in previous studies of acidic soils. Rheims et al. showed a high proportion of α-Proteobacteria from a peat bog [29], while McCaig et al. demonstrated a predominance of α-Proteobacteria in mildly acidic grasslands [30]. These results are consistent with the riparian buffers acidic soils. Of the 127 OTUs classifiable to the α- subclass, 41 and 46 OTUs belonged to the orders Rhizobiales and Rhodospirillales respectively. Clones that composed OTUs with these classifications were found in 11 of the 12 sites, with T1S3 as the exception. Species within these orders have a wide range of phenotypic characteristics, from nitrogen fixation, to methanotrophy, to phototrophy [31].
The γ-Proteobacteria were predominantly represented by OTUs classified as belonging to the Enterobacteriaceae (33 OTUs) and the Xanthomonadaceae (21 OTUs). Of particular interest was the identification of Escherichia/Shigella-related OTUs from all sites in this study. It should be noted that the source of these organisms is presently unknown. Previous studies have shown that Escherichia/Shigella sp. can be found naturally, persist, and replicate in the soil [32, 33]. Additionally, Escherichia/Shigella species have also been isolated from a number of feral species highlighting anothe potential reservoir for these microorganisms. As a result, these OTUs may not be tied to the adjacent upslope swine wastewater spray field. While 16S rDNA sequencing is not a quantitative method, the sequences that compiled these OTUs were some the most identified clones throughout the study and therefore likely represent a sizeable proportion of the microbial community in these riparian soils.
Another finding of note was the identification of OTUs in half of the libraries that were closely related to obligate methanotrophs. A number of OTUs were classified as belonging to the α-proteobacterial order Methylocystaceae, which are Type II methanotrophs. These microbes utilize methane as their sole source for carbon and energy, which potentially allows them to serve as indicator organisms for methane generation in their environment. Supporting this observation are studies demonstrating that archaeal methanogens have been isolated in high numbers from the gastrointestinal tracts of swine [34]. Thus, manure effluent may potentially serve as a reservoir of methanogens for the riparian buffer environment. Additionally, the often anoxic soil conditions found in riparian buffers are conducive to methanogenesis [35].
(ii) Acidobacteria
As a phylum amongst soil microbial communities, the Acidobacteria represent a ubiquitous and abundant group. This was reflected in our study where OTUs classified as Acidobacteria accounted for between 13 and 28% of all 12 libraries (Fig. 3), making them the second largest identified group of microbial organisms within these riparian soils behind the Proteobacteria. When dissected into subgroups, Gp1 (43 OTUs) and Gp2 (36 OTUs) accounted for the majority of the acidobacterial OTUs, followed by Gp3 (17 OTUs) and Gp6 (13 OTUs) (Fig. 8). Utilizing both clone libraries and pyrosequencing Jones et al. demonstrated that Gp1, Gp2, and Gp3 are three of the five most abundant acidobacterial subgroups within soil [36]. Lee et al. likewise demonstrated the predominance of Gp1, Gp2, and Gp3 in pH 5.5, rhizosphere soils [37]. Furthermore, Jones et al. revealed a negative correlation between the relative abundance of acidobacterial subgroups Gp1, Gp2, and Gp3 and soil pH [36]. These results may explain the high rates of these particular subgroups in the acidic soils of this riparian buffer zone. Taxonomic studies aside, the Acidobacteria remain a poorly characterized group, from both an ecological and biochemical perspective. However their abundance indicates that they play a large role within the biogeochemistry of the riparian buffer zone [38].
(iii) Planctomycetes, Verrucomicrobia, and Chlamydiae
In all 12 libraries were found a number of OTUs representing the PVC superphylum. The phyla Planctomycetes (Fig. 9) and Verrucomicrobia (Fig. 10) were each individually represented in a majority of the 12 libraries (Fig. 3), while OTUs representing Chlamydiae were found in 3 of the
sites (Fig. 10). All members of the PVC superphylum have been previously isolated from terrestrial ecosystems [39]. Both Planctomycetes and Verrucomicrobia are considered one of nine major bacterial phyla associated with soil microbial community structure [40]. The anammox bacteria, so called for their ability to anaerobically oxidize ammonia, are a monophyletic group within the Planctomycetes [41]. This study failed to find any OTUs that represented organisms capable of performing the anammox reaction. It should be noted however that these microbes are obligate anaerobes and their absence may be the product of the shallow sampling depths employed in this study; a hypothesis supported by the isolation of anammox isolates from landlocked anaerobic lagoons within the same watershed [42]. It is hypothesized that these anaerobic lagoons provide conditions suitable for the cultivation and activity of these organisms [43].
(iv) Other Phyla
OTUs classified as Actinobacteria (high G+C Gram-positive; Fig. 11) were represented in all libraries, with a range of relative abundances between 1.5 (T2S2) and 15.9% (T2S1) (Fig. 3). Firmicutes (low G+C Gram-positive; Fig. 12) and Bacteroidetes (Fig. 13) were represented by OTUs in 10 and 11 of the 12 libraries respectively (Fig. 3). Rarer OTUs that classified to the Chloroflexi/Thermomicrobia, Deferribacteres, Gemmatimonadetes, Nitrospira, OP10, Spirochaetes, and TM7 divisions were dispersed throughout the libraries (Fig. 10). For the Firmicutes-associated OTUs, approximately two-thirds (18 out of 29) classified to the order Clostridiales. For OTUs classified as Bacteroidetes, the majority (19 out of 39) classified as Sphingobacteriaceae.
Microbial Community Structure and Relationship to Denitrification
When examining the microbial community structure of the riparian buffer zone in the context of denitrification, only the phylum Proteobacteria, taken as a whole, showed a statistically significant, positive relationship with both incomplete (r2 = 0.43, P = 0.02) and total denitrification (r2 = 0.62, P = 0.002). This was not altogether surprising given that in one study, the α-, β-, and γ-Proteobacteria represented 88% of the species (out of 344 total) harboring functional denitrification genes [44]. Therefore one could reasonably predict that an increase in the relative abundance of microorganisms commonly identified with denitrification would result in a concomitant increase in denitrification potential. It should be noted however that this observation was dependent upon grouping the proteobacterial subclasses together; when taken individually, no statistically significant relationship could be found. This may relate into what Cao et al. referred to as a “functionally complementary” link between denitrifier subcommunities within an overall population, each of which become active under different environmental conditions [45]. Since the denitrification enzyme assay produces an ideal environment for denitrification activity, differences in natural behavior (i.e., temporal gene regulation) between these subcommunities may have been masked.
Additionally, spatial analysis using nonmetric multidimensional scaling (NMS) revealed that soil series played a role in the microbial community structure of the studied riparian buffer zone (Fig. 14). Sites composed of the Torhunta soil series clustered separately from sites composed of Autryville and Blanton soils. This mirrored DEA results where the three Torhunta soils exhibited the highest incomplete (iDEA) and total (tDEA) denitrification enzyme activity. Three of the four soils with high DEA rates correspond to sites located in the natural riparian zone (NRZ; Fig. 1) area. These sites are separated from the restored riparian zone (RRZ; Fig. 1) by a stream, and are closer to a field planted with row crops as opposed to the swine wastewater spray field. That eight of the nine remaining plots located in the RRZ – with the exception of T2S2 as a “hot spot” – all have low rates of both iDEA and tDEA appears to indicate that heavy loading rates of ammonia, in the form of swine manure, onto the spray field is not impacting the surrounding riparian buffer zone and resulting in high potentially high rates of nitrous oxide emissions.
Abundance of Denitrification Genes
Given this spatial connection between denitrification and microbial community structure, we further examined this relationship by looking at the abundances of genes involved in denitrification. To this aim we focused on nirK and nirS, genes which encode for separate nitric oxide reductases, and nosZ which encodes nitrous oxide reductase. Using qPCR we detected these genes in all the soils tested (Fig. 15). Relative abundances, when compared to 16S rDNA (Fig. 15) ranged from 0.02% to 1.46% (mean of 0.50%) for nosZ, 0.28% to 7.07% (mean of 2.28%) for nirS, and 0.32% to 15.64% (mean of 3.41%) for nirK. Additionally, there was a close relationship for both the total (Fig. 15; r2 = 0.42, P = 0.02) and relative (r2 = 0.92, P = <0.0001) abundances of nirK and nirS across the twelve sites studied; a pattern demonstrated in other ecosystems [46]. Relationships between environmental variables and gene abundances for nirK, nirS, and nosZ were not identified. However, relationships between total bacterial abundance with C (r2 = 0.58, P = 0.003), N (r2 = 0.57, P = 0.004), and pH (r2 = 0.32, P = 0.05) were identified. In regards to DEA, nirK, nirS, and nosZ all failed to demonstrate a statistically significant relationship, whereas total bacterial abundances exhibited a significant positive linear relationship with both incomplete (r2 = 0.57, P = 0.004) and total (r2 = 0.60, P = 0.003) DEA.
Spatial analysis demonstrated a pattern between environmental variables and denitrification gene abundances (Fig. 16) similar to that between environmental variables and microbial community structure (Fig. 14). Once again, samples taken from the Torhunta soils clustered separately from those samples collected from the Autryville and Blanton sites; this clustering of these four sites coincided with the four highest iDEA and tDEA rates found at the studied location. Additionally, the three sites located in the NRZ clustered closely together, and indicates again that, with the exception of one “hot spot” in the RRZ (T2S2), there appears to be minimal impact of heavily loading the spray field with swine manure, at least in terms of potential nitrous oxide emissions. These results indicates that while the individual genes themselves failed to demonstrate relationships with DEA, the gene abundance profiles in toto from each site could be used to infer enzymatic activity.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
Our thanks go to Dr. David Dyer, Dr. Allison Gillaspy, and the Laboratory for Genomics and Bioinformatics at the University of Oklahoma Health Sciences Center, Oklahoma City, OK, as well as Dr. Brian Scheffler and Fanny Liu of the USDA-ARS MSA Genomics Laboratory, Stoneville, MS, for DNA sequencing. We would like to thank Dr. Jarrod Miller for assistance with soil taxonomy.
The mention of firm names or trade products does not imply that they are endorsed or recommended by the U. S. Department of Agriculture over other firms or similar products not mentioned.

