All published articles of this journal are available on ScienceDirect.

Study of Genetic Evolution in Mycobacterium tuberculosis Isolates from Patients with Active Pulmonary Tuberculosis in the Iran and Belarus
Authors Info & Affiliations
Abstract
Objective:
This is the new comparative geogenetic molecular evolution research of M. tuberculosis in Iran and Belarus. Thus, we researched the genetic patterns of samples collected in the first survey of anti-tuberculosis drug-resistance by gene coding of RNA polymerase as part of the international project of on tuberculosis.
Method:
DNA extraction and amplification of rpoB gene was performed. All PCR products of gene were sequenced using the Amersham auto sequencer. For analysing phenogram has been demonstrated by method UPGMA and Neighbour-Joining. Clinical isolates (70/473) were analyzed by using sequencing gene rpoB and genotyped by program DNAMAN and MEGA.
Results:
The all data were compared with the international database of national center for biotechnology information website. Multi drug resistant of tuberculosis patient (MDR-TB) was 92% in never treated and 8% in previously treated. Mutations in rpoB gene and katG genes were showed in 95% and 84% of the MDR isolates, respectively. Two clusters were found to be identical by the four different analysis methods, presumably representing cases of recent transmission of MDR tuberculosis. The other isolates are divided in Iran into 2 groups: group A – similar to the Eastern strains (China, Taiwan) and group B – strains of another genotype. And 3 groups in Belarus: group A - Strains of the first group are more similar to the standard European and Eastern ones China and Taiwan) which diverged in the last 10 years (Genetic evolution rate), i.e. they are relatively new ones, and that is confirmed by the mutations, group B - Strains of the second group diverged earlier; they are older than the strains of the first group (16 years old- time and rate of evolution) and group C - Strains of the third group are similar to European strains and only circulate in Brest region. They are grouped separately on the phenogram and became prevalent in Iran (they are called Iranian residential strains and also is genetic analogy between group A from Iran and Belarusian isolates.
Conclusion:
This research gives a first result on genetic evolution of the M. tuberculosis strains distributing in the Iran and Belarus during the first survey of anti-tuberculosis drug-resistance and is homologies between groups A from Iran with group A from Belarus. It may aid in the creation of a national database that will be a valuable support for further studies.
INTRODUCTION
Mycobacterium tuberculosis has caused tuberculosis (TB) in humans for thousands of years [1, 2], and the World Health Organization (WHO) estimates that one third of the global population is infected with M. tuberculosis [3]; however, the bacterium has remained an enigma. The global resurgence of TB highlights the need for an improved understanding of its epidemiology and its evolutionary biologic features. Recent advances in molecular characterization of M. tuberculosis isolates, which index variation in insertion sequences [4] and repetitive genomic elements [5, 6]; have elucidated clusters of identical and closely related strain families [7-10]. These findings have provided insights into regional [11] and national [12] epidemiologic features. However, these techniques may be less suited to global population and evolutionary analyses, and integrating information obtained from different approaches is complex [4, 13]. Genomic comparisons have identified genetic variation for population screening; however, these analyses are limited to those sites that vary between the compared genomes and are potentially misleading [2, 3, 14-16]. Nucleotide sequences provide robust, portable, and comparable data for studying population variation. The mutational processes that generate this variation are understood, and sequence data have been successfully used in the study of bacterial epidemiology, population biology, and evolution [17]. The complete genome sequences (16–19) provide access to all regions of the chromosome and facilitate such studies. However, high-throughput gene sequencing of structural genes [20] and host immune system protein targets (21) in M. tuberculosis isolates indicated low levels of sequence diversity. In M. tuberculosis, resistance to drugs (rifampicin and Isoniazid) occurs because of chromosomal in certain genes that reported by investigators in the katG and rpoB gene [21-23].
Although extensive genomic sequencing was performed in both studies, comparable sequence data were obtained on a limited number of highly selected isolates.
Ongoing research is focused on identifying the factors responsible for the worldwide spread of the W-Beijing strains and their ability to adapt and enhance their pathogenisity or virulence. Identifying a possible mechanism for increased adaptation of these bacteria to the human immunologic host defense system or human interventions such as anti-TB treatment is of the utmost importance. Such mechanisms may indicate how the bacterium adapts to the host, a prerequisite for an enhanced accumulation of chromosomal associated with resistance.
Despite an unusually high degree of relatedness at the genetic level, the five species comprising the M. tuberculosis complex differ in their host range, their phenotypes and ability to cause disease in humans. Variations in virulence are even observed, in animal models of the disease, between members of the M. tuberculosis species. Comparative genomics has been applied to the M. tuberculosis complex in an attempt to understand the molecular basis for these differences.
The genome sequences of the paradigm strain, H37Rv, and a recent clinical isolate, CDC1551, comprise 4.41 and 4.40 Mb, respectively, and harbour ~4,000 genes coding for proteins. In contrast, the genome of M. bovis strain AF2122/97 at 4.34 Mb is significantly smaller as the result of a series of deletion events that have removed ~80 genes.
The genetic evolution characterization of the isolates selected during this study was not carried out and there is no information on the major distributing clades of M. tuberculosis. The change of nucleotide involved in antibiotic resistance and the question of whether the Beijing family of the M. tuberculosis complex had spread into Iran have not been studied until now and were not comparative between European country and Asian Country. Molecular epidemiological studies have demonstrated the widespread of this family and its association with drug-resistance [6, 7, 9].
In recent years, the genetic typing of M. tuberculosis complex (MTC) strains has been widely used to support conventional epidemiological investigations of TB outbreaks and as a tool for studying transmission dynamics.
In this work, has been investigated the genetic evolution of isolates selected in the first survey of anti-tuberculosis antibiotic-resistance by analyses rpoB fragment sequence in the MDR strains from Belarus and Iran and has been identified homologies between groups A from Iran with group A from Belarus.
MATERIALS AND METHODS
Mycobacterial Strains
A total of 895 M. tuberculosis strains were isolated from the sputa of patients with active pulmonary tuberculosis and collected from the different region of Iran and Belarus that 70 isolates were recovered resistance to rifampicin and 66 isoniazid-resistant. And 32 isolates were multi drug resistant (MDR), from Feb to December 2008. All 283 tuberculosis patient cases had proven registration of clinical diagnostic examination, such as chest X-ray, PPD, cough, weight loss, gender, etc. All isolates were cultured on Lo¨wenstein-Jensen solid medium and grown colonies were identified to the species level using TCH (2-thiophene carboxylic acid) and PN99B (paranitrobenzoic acid) selective media using Centers for Disease Control and Prevention (CDC) standard biochemical procedures. M. tuberculosis reference strains CDC1551 and H37Rv and four susceptible isolates of M. tuberculosis were used from the banked strain collection of the Pasteur Institute of Iran. M. tuberculosis strain 210 is a susceptible reference isolate from the M. tuberculosis bank at the Belarusian Research Institute of Epidemiology and Clinical Microbiology and was also used as an internal negative control [5].
Susceptibility Testing
Antimicrobial drug susceptibility testing (AMST) was performed using the CDC standard conventional proportional method: rifampin (40 _g/ml), isoniazid (2 _g/ml), ethambutol (2 _g/ml), ethionamide (20 _g/ml), streptomycin (4 _g/ml), and kanamycin (20 _g/ml) were used in slants and in addition to breakpoint concentrations for isoniazid (0.1 _g/ml) and rifampin (2.0 _g/ml) in the Bactec system (10). Four sensitive M. tuberculosis isolates and an H37Rv strain were used as negative controls. Mutations in the rpoB gene were identified from 41 rifampin-resistant isolates by sequencing methods (see below), and AMST was performed following sequencing to confirm resistance using different concentrations of rifampin (50, 75, and 100 _g/ml) through the slant proportional method [8] and also using BACTEC system (Becton Dickinson company in tuberculosis laboratory of Institute of pulmonology and tuberculosis in Minsk, Belarus.
PCR Amplification
DNA was extracted and purified using the Fermentas DNA extraction procedure (K512). M. tuberculosis reference strains CDC1551 and H37Rv and four sensitive isolates of M. tuberculosis from the banked strain collection of the Massoud and Zia Zrifi Laboratories were used as negative controls. A 411-bp fragment of the rpoB gene and 209-bp fragment of katG was amplified by PCR with primers rpoBF (5-TACGGTCGGCGAGCTGATCC-3) and rpoB-R (5-TACGGCGTTTCGATGAACC-3) and katG-F 5-GAAAC-AGCGGCGCTGATCGT-3, katG-R 5-GTTGTCCCATTT-CGTCGGGG-3(22). PCR was carried out in 50 μL of a reaction mixture containing 50 mM KCl, 10 mM Tris (pH 8.0), 1.5 mM MgCl2, 5 μM of deoxynucleoside triphosphates (dNTPs), 1U Taq polymerase, 20 pmoles of each set of primers, and 6 μM of chromosomal DNA (24). For rpoB fragment Samples were then subjected to one cycle at 94°C for 5 min, followed by 36 cycles at 94°C for 1 min, 57°C for 1 min, 72°C for 1 min, and a final cycle at 72°C for 10 min to complete the elongation of the PCR intermediate products. For katG the following thermocycler parameters were used: initial denaturation at 94°C for 5 min; 42 cycles of denaturation at 94°C for 1 min; primer annealing at 57°C for 1 min; extension at 72°C for 1 min; and a final extension at 72°C for 10 min. PCR products were then run on 2% agarose gels and examined for the presence of the 411-bp and 209 bp band after ethidium bromide staining. The DNA Extraction were performed on agaros using Sigma Kit (124K6083) and the products were checked and purified on the gel electrophoresis and purified rpoB and katG segment were amplified. The PCR product was amplified, purified again, and controlled on the electrophoresis gel. The final purified mycobacterial DNA was used for sequencing.
DNA Sequencing of the rpoB Fragment
Sequencing of 411-bp fragment were done using the same forward and reveres primers; 33 cycles of denaturation at 94°C for 30min; primer annealing at 54°C for 30 sec; extension at 72°C for 90 sec, A 411-bp fragment of the rpoB gene extracted from tuberculosis strains were sequenced by Amersham auto sequencer and Amersham Pharmacia DYEnamic ET Terminator Cycle Sequencing Premix Kits. Alignment of the DNA fragments (rpoB) was carried out with the help of MEGA software (Gen bank_ PUBMED/BLAST).
DNA Sequencing of the katG Fragment
Sequencing of 209-bp fragment of katG gene was amplified by PCR using forward and reveres primers; 33 cycles of denaturation at 94°C for 30sec; primer annealing at 48°C for 45 sec; extension at 60°C for 4 min. The katG gene fragments of tuberculosis isolates were sequenced using the Amersham auto sequencer and Amersham Pharmacia DYEnamic ET Terminator Cycle Sequencing Premix Kits.
Data Analyzing of DNA Sequencing
The 411-bp fragments of the rpoB gene were amplified by PCR using the forward or reverse primers mentioned above using the following conditions: 33 cycles of denaturation at 94°C for 30 s, primer annealing at 57°C for 30 s, and extension at 72°C for 90 s. The rpoB gene fragments were sequenced by using the protocol of the DYEnamic ET dye terminator kit (MegaBACE) and a MegaBACE 4000 DNA sequencer (Amersham Biosciences [currently GE Healthcare], Piscataway, NJ). Alignment of the DNA fragments (rpoB) was carried out using MEGA and DNAMAN software and was compared with the standard M. tuberculosis reference strains CDC1551, H37Rv, and 210. The Blast2 sequencing program (NCBI) was used for DNA sequence comparisons (http://www.ncbi.nlm.nih.gov/BLAST/). Alignment of the DNA fragments (rpoB) was carried out with MEGA 3.1 software (www.megasoftware.net/), and data were analyzed and edited with DNAMAN software (Lynnonon Biosoft, Quebec, Canada).
In this study, all smear microscopy, acid-fast bacilli (AFB) culture, susceptibility testing, and PCR assays were performed at the mycobacteriology department of the Pasteur Institute of Iran, and the DNA sequencing analysis was conducted at the Belarusian Research Institute of Epidemiology and Microbiology, Clinical Microbiology Department. The obtained data were analyzed and edited with DNAMAN programs.
Mutations Spectrum and Frequency Analysis
DNAMAN is a sequence analysis software package for IBM compatible computers with Microsoft Windows 95/98 or NT/2000 systems. This package provides effective and convenient tools for molecular geneticist to deal with frequently used analyses in research. It contains text editor for fragments sequence alignment. The text editor is identical to WINDOWS WordPad. The editor allows deleting or pasting any text or sequence to files [24].
Cluster Analysis by Neighbor-Joining Method
The basic unit of data used in Neighbor-Joining (Nearest Neighbor) cluster analysis is the homologies coefficient derived from any one of the homologies analysis formula. A homologies matrix derived from sequential pair wise comparisons of two most similar operable taxonomic units (OTU) or clusters of OTUs is used to construct a phenogram. This phenogram is setup with the distance matrix using the Neighbor-Joining method. Phylogenetic evolution tree demonstrated related homologies between any two sequences in a multiple alignment [24].
Unweighted Pair-Group Method Using Arithmetic Averages
Unweighted pair-group method using arithmetic averages (UPGMA) is one of the most frequently used cluster analysis methods an is widely applied in the construction of phenograms based on electrophoretic fingerprint patterns, hybridization matrices, and nucleic acid sequences. With UPGMA, an OTU, or every OTU in a cluster of OTUs is joined to a known cluster on the basis of the average (mean) distance between all combinations of pairs of OTUs. Thus, UPGMA calculates the average distance from one cluster of OTUs to a comparison cluster, where each OTU in the cluster is given equal weight. This phenogram is setup with the distance matrix using the UPGMA method. The matrix can be built up only with Observed Divergence method. Phenogram shows related homologies between two sequences or groups [24].
RESULTS AND DISCUSSION
Mycobacterial Strains and Susceptibilities
In this study, we had 286 available M. tuberculosis isolates, of these, 59 (20.8%) had never been treated for tuberculosis and the other 27(9.5%) had previously received treatment. Of the 283 isolates, 211 had sensitive to all four evaluated antibiotics and 72 (25.4%) demonstrated resistance to rifampicin with other drugs or isoniazid (n=68) with other drugs.
The rate of multi drug resistant (MDR) was 12.1% (n=34). Most of the patients with drug-resistant isolates (74/ 90-82%) were aged between 15 and 54 years.
Drug Susceptibility
All 72 isolates of rifampicin- isoniazid resistant examined were resistant to rifampicin (97%), isoniazid (91%), streptomycin (90%) and 18 isolates (48%) were resistant to etambutol. In this research has been identified two strains mono-resistance to rifampicin and isoniazid resistant was isolated for 4 isolates.
Analysis for rpoB Gene – Iranian Isolates
60 mutations and 13 micro deletions were identified in 29 rifampicin-resistances MBT (85%). In 5 rifampicin-resistances MBT isolates (15%) no mutations were found in the core region of the rpoB gene. Of 60 found mutations 6 silent (8.3%) and 54 (91.7%) were missense. Most of detected deletions were identified in codon 510 GAG/_AG (12.5%). All silent mutations were localized in codon 507, missense mutations revealed 23 types of amino acid substitutions. Most frequent mutated codons in Iranian strains were 523 GGG/GG_, GGG/GCG and 526 CAC/TAC, CAC/CGC, CAC/AAC, CAC/TTC, CAC/CAA, CAC/_GC (six types of mutations). Mutations in codons 510, 507, 531 were observed in 27%, 24%, and 21% of isolates and correspondingly Mutations in codon 523 resulted in Gly523Ala replacement and in codon 531 Ser531Leu and Ser531Phe. We observed 6 alleles in codon 526, 3 alleles in triplets 507, 508, 513. In 6 strains (18%) harboured single mutations placed in codons 526, 510, while isolates with multiple mutations revealed double 34%, triple 22% and quadruple 3% of the strains. 12% of strains harboured 5 mutations (Fig. 1).
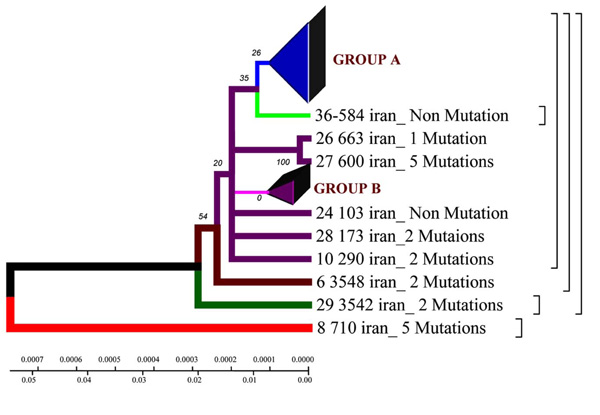
Phylogenetic geogenetic phenograms of M. tuberculosis isolates collected from Iranian tuberculosis patients within the time period of 70 years by Neighbour-Joining method. * Frequency of mutation has been indicated for isolates in the branches.

Distribution of geogenetic groups of M. tuberculosis on the territory of Iran.
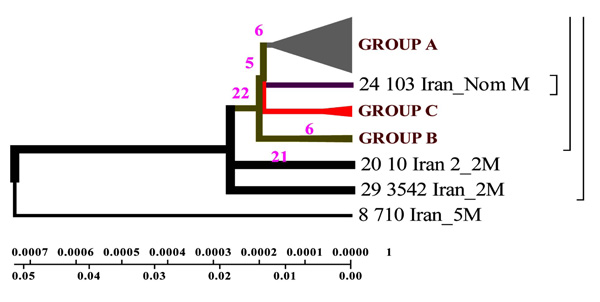
Geogenetic phenogram constructed by Minimum method for Iranian strains. M= Mutation, In the this figure mutation has been demonstrated for Iranian Isolates.

Evolutionary genetic phenogram of M. tuberculosis of Belarusian isolates constructed by UPGMA method.
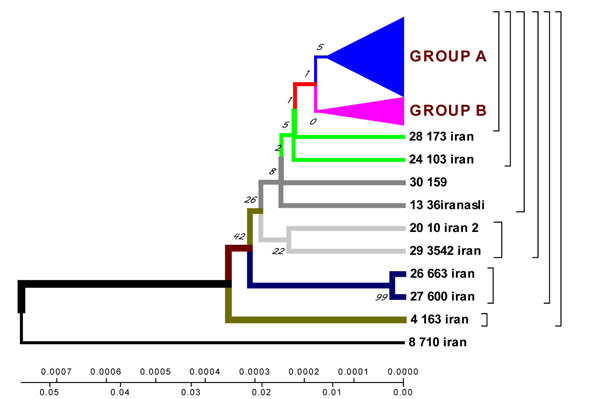
Geogenetic phenograms and their classification obtained by UPGMA method for Iranian Isolates.

Distribution of geogenetic groups of M. tuberculosis on the territory of Belarus.
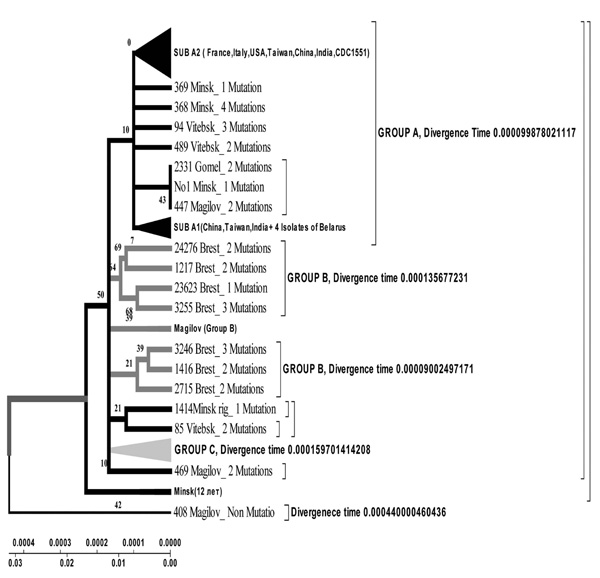
Evolutionary geogenetic phenogram of 44 Belarusian M. tuberculosis isolates constructed by Neighbour-Joining method. * Frequency of mutation has been indicated for isolates in the branches.
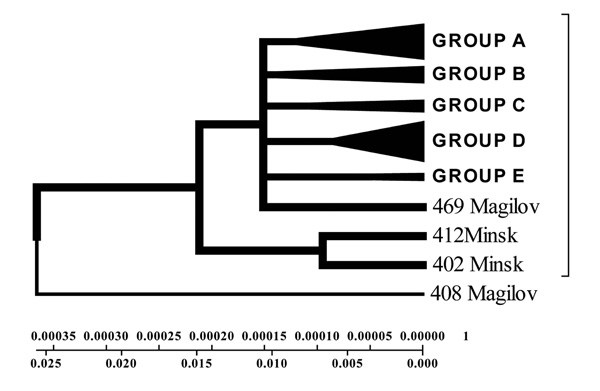
Geogenetic Phenogram constructed by Minimum method with the use of Belarusian isolates sequences.
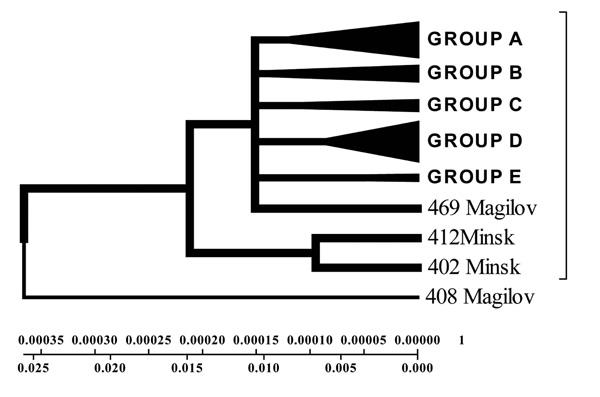
Geogenetic Phenogram and classification of M. tuberculosis isolates by UPGMA method.
Analysis for katG Gene – Iranian Isolates
All 28 samples were cultured and identified as M. tuberculosis by PCR method that reveals 49 mutations in all stains. From 26 isoniazid-resistance 2 strains showed no mutation and in 21 strains mutations were observed in codon 315, revealing three types of mutations consist of AGC→ACC (Ser →Thr) (80%), AGC→AGG (Ser→Arg) (5%) and AGC→AAC (Ser→Asn) (15%). One type of mutation obtained in codon 299 indicating GGC→AGC and changes in amino acid Gly→Ser. In codon 311(18%) (katG) only one base change were obtained Asp→Tyr (GAC → TAC) in nine strains demonstrate a nonsense mutation. Furthermore only one mutation observed in codons 311,299 and 322. And in 12 strains one mutation in codon 315(42.8 %), 7 strains 2( 25%), 5 strains 3(17.8 %) and 2 isolates 4 mutations (7.1 %) were obtained respectively (Fig. 2).
Analysis for rpoB gene - BelarusianIsolates
16 different types of nucleotide change has been identified for Belarusian isolates and from these, 14 ones in four M. tuberculosis isolates in codons 520 (CCG→CCC)-2, 521 (CTG→TTG), 508 (ACC→ACG)-2, 523 (GGG→GGA), (GGG→GGC)-4 and 510 (CAG→TAG)-4 were identified to the silent mutations. All mutations showed were point ones except the one in codon 531 which is a substitution TCG→TGC. The most frequently mutated codons were 510, 526 and 523 with corresponding frequencies 47.7%, 40.9% and 38.6%. Of forty five mutated isolates eleven (27.7%) had one mutation, twenty three (52%) – two mutations, seven (16%) – three mutations, four (7%) – four ones. Of the 44 Iranian rifampin-resistant isolates, 31 randomly selected isolates were chosen based on the WHO-defined clinical symptom definition (35). Of these 31 rifampin-resistant isolates, 22 were isolated from the sputa of patients with primary infections and 9 (41.30%) were from that of patients bearing secondary cases. Among the strains mentioned double mutations were detected of which three in codons 531 (TCG→TTG) and 510 (CAG→GAG). Double mutation was also observed in codons 531 (TCG→TTG), 523 (GGC→GCG), 512 (AGC→GCC) and 507 (GGC→GCC). One strain had thee silent mutations in codons 520 (CCG→CCC), 507 (GGC→AGC) and 510 (CAG→AAG). The research accomplished showed that one mutation was more frequently observed in codons 526, 531 (36%, 36%); two ones – in codons 526, 510 (52%, 52%). Three or four mutations were detected in codons 510 (70%). To study the prevalence of mutations in the Republic of Belarus comparison was made depending on the region of residence. According to the result in all regions of Belarus two mutations are found with the same frequency. Four mutations in rpoB gene were only detected in Minsk (2.27%) and Minsk region (4.54%).
Analysis for katG Gene - BelarusianIsolates
In the four isolates of all isolates that were sensitive to isoniazid any mutation were not identified in the region 209 bp of katG gene. Nucleotide change were detected in affected codons of PCR product segment 209 bp of katG gene, 305, 306, 307, 314, 316, 321, 328, 315, 316,357, 454,463 and 309. Four types of mutations were demostrated in codon 315: AGC→ACC (n=36)36%, AGC→AGG (n=1)0.9%, AGC→AAC (n=2)1.8%, AGC→GGC (n=1)0.9%. One type of mutation was showed in codon 316: GGC→AGC (n=16)%, four types of mutations were identified in codon 309: GGT→GGT (n=7)%, GGT→GCT (n=4)%, GGT→GTC (n=3)%, GGT→GGG (n=1)%. Mutations in codon 309 revealed 34%, in codon 316 – 37%, other types of mutations 29% of all detected mutations. Among all isolates (n=40) 95% of mutation were demostrated in codon 315 (four type mutation). For two isolates 2 types of mutations were seen in codon 357 GAC→CAC and GAC→AAC, in addition two mutations were also observed in codon 463 CGG→CTG and 454 GAG→CGA. In all 42 strains, one mutation 21.5%, 16 – two mutations38%, 9 - three mutations21.5%, four – 4 mutations9.5% and 4 – five mutations9.5% were determined. Two isolates have not any mutations in kat G gene. Three silent mutations were detected in codons: 306 (CCG→CCC), 309 (GGT→GGG) and 314 (ACC→ACG) these silent mutations revealed no effect on the susceptibility pattern tested strains.
EVOLUTIONARY GENETIC ANALYSIS OF M. TUBERCULOSIS ISOLATES FROM IRAN
Sequencing of 81 bp fragment of rpoB gene was carried out for 34 strains. From of the sequences obtained, a phenograms was constructed by UPGMA and Neighbour-Joining methods in DNAMAN software.
Homologic phenogram was constructed by UPGMA method using distance matrix. According to the phenogram, the Iranian strain 710 is closely related to the strains from Taiwan, China and India (BLAST software). The other strains are supposed to have originated from this one.
Phylogenetic phenogram. With the help of Neighbor-Joining method the data obtained confirm the results of Neighbor-Joining method for the Iranian strain 710 (Fig. 1).
According to the phenogram one strain from the town of Zabol is older than the rest of the strains (it is about 77 years old), and it was considerably changed after its appearance in Iran and was resistant to anti tuberculosis drugs (Fig. 4,9). This strain contains five mutations. When it appeared in Iran it possessed drug resistance. The other strains similar to each other are located on the same branch of the phenogram (Fig. 1).
The other strains are divided into 2 groups: group A – similar to the standard and Eastern strains (China, Taiwan), they are about 10 years old. These strains were changed in Iran and acquired mutations (Fig. 4.9); group B – strains of another genotype. They are grouped separately on the phenogram and became prevalent in Iran (they are called Iranian residential strains) (Fig. 2).
DNAMAN software allowed to obtain general classification of M. tuberculosis collected from the patients from different regions of Iran. In the phenogram constructed by Minimum method all strains are divided into three genetic groups and in each of them there are closely related strains (Fig. 3).
Strains of group A are similar to the standard ones, and formed not long ago; group B comprises two strains of earlier origin than the others, and it’s obvious that they were changed after their appearance in Iran; group C comprises four strains which appeared not long ago.
According to the phenogram constructed by UPGMA method all strains are divided into two genetic groups. Some of the strains differ in genotype and divergence time and are located separately (figures 4.12 – 4.13). 60% of Iranian strains diverged as a result of mutations from the same M. tuberculosis strain 710 similar to the strains from China, Taiwan and India. 20% of the strains had the highest homologies with the ones from Taiwan. One isolate (from Zabol region) 710 is related evolutionary to ancestral M. tuberculosis family. In this sample there are no mutations in codon 531. Strain 163 located separately and the number of strains combined in one group originated from it. Four isolates belong to the standard M. tuberculosis strains (H37RV, CDC1551), and on of these four is similar to Indian strain 3062 Iran (Fig. 4).
23 isolates are combined in groups and have Iranian origin. Five isolates diverges from the primary sample: one from Tehran, one from Kurdestan, three ones are similar to the strains from Taiwan (TMB 522). All the strains from Iranian patients are divided into two genetic groups: A – similar to Eastern strains (Taiwan, China, India, Japan, and Korea), they are about 11 years old; B – Iranian residential strains which appeared more than 20 years ago (Fig. 5).
In the cause of study of mutations in rpoB gene in the strains from Iranian patients the most frequent changes were found in codons 523 and 526. In the isolates from the patients with primary tuberculosis (12 strains) the changes were detected in codons 526 (28.5%), 523 (21.4%), 510 (14.2%). In the isolates from the patients with secondary tuberculosis (22 strains) mutations were found in codons 526 (13.5%), 523 (23.7%) and 510 (11.8%).
In Iranian M. tuberculosis strains in katG gene the most affected codons were 315 and 311. In the strains from the patients with primary tuberculosis the changes were detected in codons 315 (25%), 311 (0.9%), 299 (0.9%). In the strains from the patients with secondary tuberculosis mutations were found in codons 315 (36.8%), 299 (18.4%), 311 (21%). According to the phenograms constructed for rpoB gene the strain 710 from the town of Zabol is more closely related to the Chinese strains. The other strains are supposed to have diverged from this strain. Iranian strain 710 has the greatest homologies with the strains from China, Taiwan and India. Strain 710 from Iran has the highest homologies of its genome with the strains from China, Taiwan and India. It appeared about 77 years ago.
Using Neighbour-Joining method evolutionary phenograms were obtained for Iranian strains which were divided into two genetic groups: group A – strains more related to Eastern ones (Taiwan, China, India, Japan, and Korea); group B – Iranian residential strains.
Evolutionary phenograms constructed for the first time for M. tuberculosis strains collected in Iran show that strain 710 of rpoB gene collected from the town of Zabol is closely related to the strains from China, Taiwan and India. The other strains are supposed to have diverged from this one. All strains analysed collected from Iranian patients are divided into 2 genetic groups: group A – similar to Eastern strains (Taiwan, China, India, Japan, Korea) (they are about 11 years old); group B – Iranian residential strains which appeared about 23 years ago.
Construction of evolutionary phenograms using sequences of M. tuberculosis resistance genes collected in Iran and bioinformatics analysis are recommended to study epidemic process of tuberculosis.
EVOLUTIONARY GENETIC ANALYSIS OF M. TUBERCULOSIS ISOLATES FROM BELARUS
411 fragment of rpoB gene sequences were used to construct a phenogram with the help of MEGA and DNAMAN software.
Phylogenetic phenogram. With the help of nucleotide sequencing methods precise description of small gene fragments can be obtained; with the help of other methods of molecular strain typing larger genome fragments can be compared, but finally the comparison should be fulfilled by means of homologies and difference evaluation. For this purpose a distance matrix should be constructed by Neighbour-Joining method. An assumption is made about independent evolution of characters. In the cause of bootstrapping other analysis functions can be used. The data obtained with the help of this software confirm the results of Neighbour-Joining method for the strain 408 isolated from the patient from Mogilev region.
Isolates collected from the tuberculosis patients from Belarus were branched into 3 genetic groups. The first one is closer to European isolates (from Italy, France, Finland), the second one – to Central Asia (from Russia, China, Taiwan), the third one – Belarusian resident (Fig. 7).
According to the phenogram the isolates selected from Mogilev city patients is essentially older (44 years old). Within this period it was not considerably changed after its appearance in Belarus. The other isolates with the help of bioinformatics analysis has been branched into 3 main geogenetic groups:
- Isolates of the first group were more similar to the isolates from China and Taiwan which diverged in the last 10 years (evolution genetic rate), i.e. they were relatively new ones, and that is confirmed by the nucleotide changes (Fig. 6, 7).
- Isolates of the second group diverged earlier; they were older than the isolates of the first group (16 years old- rate of evolution genetic) (Fig. 7).
- Isolates of the third group are similar to European isolates and only circulate in the Brest city.
All the other isolates were very similar to each other and located on the same branch (Fig. 8). With the help of bioinformatics software general classification of M. tuberculosis isolated from the tuberculosis patients from different cities of Belarus was obtained. In the phenogram constructed by Minimum method all isolates were divided into five branches, and in each branch there were neighbouring isolates (Fig. 8).
According to the phenogram constructed isolate 408 from Mogilev city was closer in its primary structure to the isolate from Russia and China (Fig. 6-9).
According to unweighted pair groups' method with use of arithmetic average (UPGMA) it was obvious that the isolates from Mogilev was older, and the other ones associated with each other depending on the homologies level of the fragments examined (Fig. 9). It is evident that the Brest isolates differ from the rest ones and probably originate from Western Europe and represent a separate cluster (Fig. 9).
Five isolates made up a separate branch from the first one: one strain from Mogilev city, another – from Vitebsk city. Sequences of 3 isolates were close to the ones of the isolates from Italy, India, and Taiwan. Sequences of four isolates correspond to the ones of the standard strains H37RV and CDC1551. One isolate from the city of Minsk completely corresponds to the standard isolates. In this study, we identified different geogenetic isolates from Belarusian tuberculosis patients that distributed in the cities of Belarus that similar to isolates from India, Taiwan, Italy and China (Fig. 2-4).
With the help of Neighbour-Joining method evolutionary genetic phenograms were obtained for Belarusian isolates which were divided into three geogenetic groups: A – isolates more similar to Asian isolates (Taiwan, China); B – Belarusian strains; C – European strains (Fig. 6, 7).
According to the phenograms obtained by UPGMA method strains were divided into three geogenetic groups: A – closely related to Eastern strains (Taiwan, China); B – Belarusian residential isolates; C – related to European isolates (Fig. 9).
Evolutionary genetic phenograms constructed for the first time for M. tuberculosis isolates selected in Belarus show that isolates 408 of rpoB gene collected from the tuberculosis patient from Mogilev city was more related to the isolates circulating in Russia and China. The other isolates were divided into three geogenetic groups (appeared about 9 years ago- rate and time of evolution - Fig. 6-9).
Construction of evolutionary genetic phenogram using sequences of M. tuberculosis resistance genes collected in Belarus - Iran and bioinformatics analysis are recommended to study epidemic process of tuberculosis.
In the final inclusion, we have resulted that from all isolates in Iran and Belarus is genetic analogy evolution in the group A - closely related to Eastern Asian strains and this analogy was most in the isolates from capital of countries. This result has been indicated that Asian strains circulated between countries via person in which contaminated by M. tuberculosis and is distributing speedily.
AUTHORS' CONTRIBUTIONS
LA conceived the study, carried out the molecular genetic studies, analyzed the data and drafted the manuscript; RA, ME, AG and AVdeS participated in the design and carried out the first survey of anti-tuberculosis drug-resistance, analyzed the data, and provided the clinical isolates for molecular study. CR and JR carried out mycobacteriological diagnostics, isolation of clinical isolates, identification, drug susceptibility tests and provided information about the clinical isolates; CL participated in the genotyping studies; AC assisted with data entry, conducted the bioinformatics programming and statistical analysis; MAL and MJR provided technical help in the conservation of the strains; CS and NR carried out the phylogeny reconstruction studies, participated in the identification and designation of the SITs and also helped draft the manuscript; CM participated in the design of the study, and provided critical comments for the manuscript; SS conceived the study, and participated in its design, coordination of the investigation, and helped to draft the manuscript for consideration for publication. All authors contributed to the study, read and approved the final manuscript.

