All published articles of this journal are available on ScienceDirect.

Genomic Analysis Reveals Versatile Organisms for Quorum Quenching Enzymes: Acyl-Homoserine Lactone-Acylase and -Lactonase
Abstract
Microbial virulence and their resistance to multiple drugs have obliged researchers to look for novel drug targets. Virulence of pathogenic microbes is regulated by signal molecules such as acylated homoserine lactone (AHL) produced during a cell density dependent phenomenon of quorum sensing (QS). In contrast, certain microbes produce AHL-lactonases and -acylases to degrade QS signals, also termed as quorum quenching. Mining sequenced genome databases has revealed organisms possessing conserved domains for AHL-lactonases and –acylases: i) Streptomyces (Actinobacteria), ii) Deinococcus (Deinococcus-Thermus), iii) Hyphomonas (α-Proteobacteria), iv) Ralstonia (β-Proteobacteria), v) Photorhabdus (γ-Proteobacteria), and certain marine gamma proteobacterium. Presence of genes for both the enzymes within an organism was observed in the following: i) Deinococcus radiodurans R1, ii) Hyphomonas neptunium ATCC 15444 and iii) Photorhabdus luminescens subsp. laumondii TTO1. These observations are supported by the presence motifs for lactonase and acylase in these strains. Phylogenetic analysis and multiple sequence alignment of the gene sequences for AHL-lactonases and –acylases have revealed consensus sequences which can be used to design primers for amplifying these genes even among mixed cultures and metagenomes. Quorum quenching can be exploited to prevent food spoilage, bacterial infections and bioremediation.
INTRODUCTION
The advent of antibiotics in the beginning of twentieth century gave a new lease to human health and longevity to life span. Ironically, usage of antibiotics to counter the various diseases caused by pathogenic microbes has been proving quite ineffective during the last decade or so. The situation is turning alarmingly grave because microbes have become increasingly resistant to almost all the antibiotics including those which had been approved quite recently [1, 2]. Pharmaceutical companies find it uneconomical to be in this evolutionary race with multiple drug resistant microbes [3-6]. In fact, microbes have evolved defence mechanisms against antibiotics by exploiting their large reservoir of genetic variability [6]. The needs are to search new drug targets [7, 8] and /or attenuate bacterial pathogenicity [9] by preventing dissemination of genes conferring resistance and those responsible for their virulent characteristics [3] and design drugs accordingly.
Among the various microbial pathogens, those notorious for causing high morbidity and mortality among living beings initiate the infection process within specialized structures – biofilms, which enable microbes to withstand adverse environmental conditions, such as the host defence components and drugs including antibiotics used for treating the disease [10, 11]. Most pathogenic microbes develop it in a density dependent manner by a process termed as quorum sensing (QS) [12], which is regulated by the signal molecules released in their environment [13]. In Gram-negative bacteria, the most widely reported QS system is LuxR/I-type, where signals such as acylated homoserine lactones (AHLs) regulate the expression of virulence factors like elatase and pycocyanin production by Pseudomonas aeruginosa [14], antibiotic production by Erwinia carotovora, pigment and antibiotic production by Serratia liquefaciens [15]. The AHL-type pheromones differ in their specificity due to variations in acyl chain length, degree of saturation and substitutes at the third carbon of the lactone ring [16]. More than 70 bacterial species produce AHL type QS [15, 17].
In contrast to AHL mediated QS, which allows bacteria to compete and dominate the bacterial community, there are others which have developed mechanisms to degrade these signals. The process of interference with QS, known as quorum quenching (QQ), has been reported among prokaryotes and a few eukaryotic organisms [18, 19]. Bacteria are known to produce AHL-degrading enzymes such as AHL-lactonases and AHL-acylases. Lactonases hydrolyze the lactone ring of AHL and are found in numerous Bacillus spp. [20, 21]. Heterologous expression of aiiA (encoding for AHL-lactonase) from Bacillus spp. in Pseudomonas, Burkholderia and Erwinia resulted in marked decrease in their QS signal molecules [22-24]. AHL-acylases break the amide linkages of the AHLs and have been reported to be produced by bacteria such as Ralstonia spp. [25, 26] and Pseudomonas spp. [27-29]. Rhodococcus erythropolis W2 presents an interesting scenario, as it possess both types of AHL-degrading enzymes [30-32]. Recently proposals have been floated to use QS signal molecules as potential drug targets to attenuate the virulence of pathogenic bacteria and use QQ to design novel drugs [25]. The large reservoir of information available in sequenced genomes of many organisms encouraged us to search for potential organisms [4, 33-35], which may possess either or both the enzymes for usage as potential drugs or as supplement to antibiotic based treatments.
MATERIALS AND METHODOLOGY
Phylogeny of Acyl-Homoserine Lactone Degrading Enzymes
From the literature available in public domains, we collected protein sequences for AHL-lactonase from Bacillus sp. SB4 (Accession No. AAR85482.1) and AHL-acylase from Ralstonia sp. XJ12B (Accession No. AAO41113.1) from NCBI Protein database. The nucleotide sequences of corresponding protein sequences for the MEME signature analysis were downloaded from the NCBI Genbank. The conserved domains of these enzymes from reference organisms have been presented in Fig. (1). We used BLASTP for the similarity searches and NCBI Conserved Domains [36, 37], for conserved domain search. The selected sequences were analyzed to get similar sequences and Conserved Domains of each BLAST hit. We selected parameter for Max target sequences in BLASTP as ~250 hits (BLOSUM 62 Matrix), as it gives results with large number of sequences from same species.
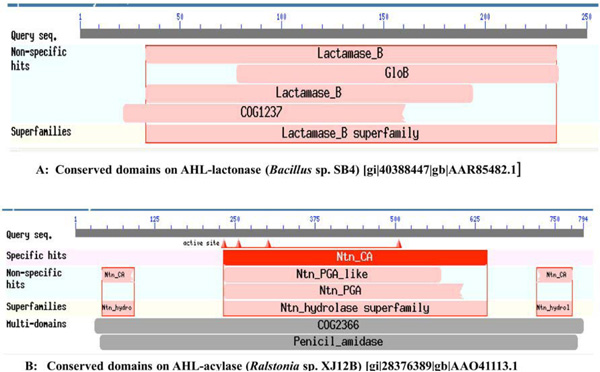
A. Conserved domains on AHL-lactonase (Bacillus sp. SB4) [gi|40388447|gb|AAR85482.1], B. Conserved domains on AHL-acylase (Ralstonia sp. XJ12B) [gi|28376389|gb|AAO41113.1].
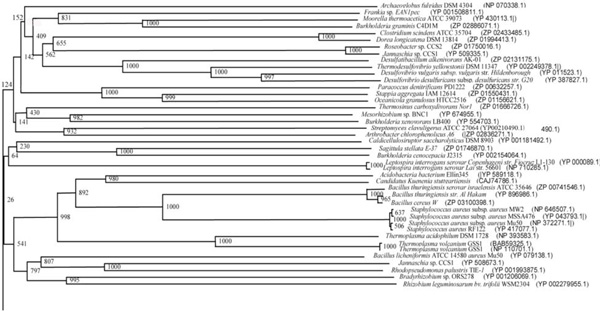
Phylogenetic tree (Upper half) based on protein sequences of organisms containing conserved domains with respect to Bacillus sp. SB4 AHL lactonase as reference (For Taxonomic details refer to Additional file 3). A Neighbor-Joining analysis with Jukes-Cantor correction and bootstrap support was performed on the protein sequences. Bootstrap values are given at nodes, 1000 bootstrap replicates were run. The leaf contains Accession number and name of the organism.
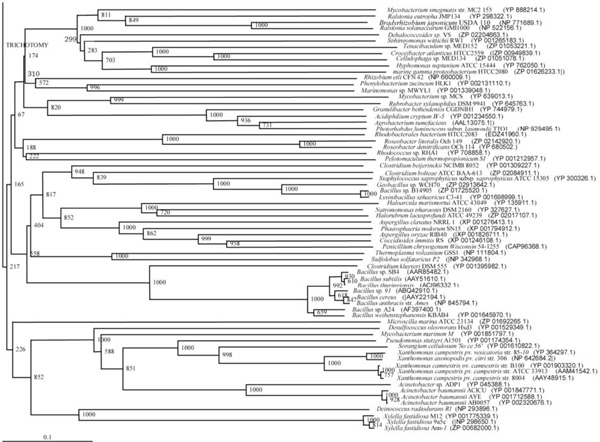
Phylogenetic tree (Lower half) based on protein sequences of organisms containing conserved domains with respect to Bacillus sp. SB4 AHL lactonase as reference (For Taxonomic details refer to Additional file 3). A Neighbor-Joining analysis with Jukes-Cantor correction and bootstrap support was performed on the protein sequences. Bootstrap values are given at nodes, 1000 bootstrap replicates were run. The leaf contains Accession number and name of the organism.
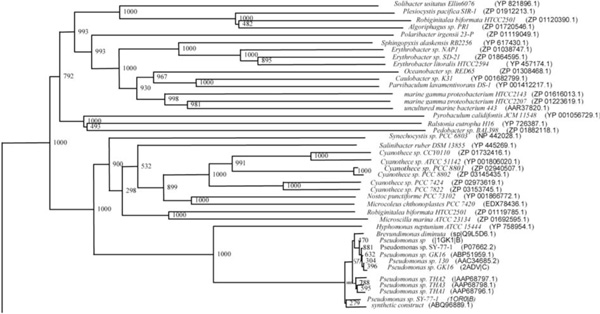
Phylogenetic tree (Upper half) based on protein sequences of organisms containing conserved domains with respect to Ralstonia sp. XJ12B AHL acylase as reference (For Taxonomic details refer to Additional file 3). A Neighbor-Joining analysis with Jukes-Cantor correction and bootstrap support was performed on the protein sequences. Bootstrap values are given at nodes, 1000 bootstrap replicates were run. The leaf contains Accession number and name of the organism.
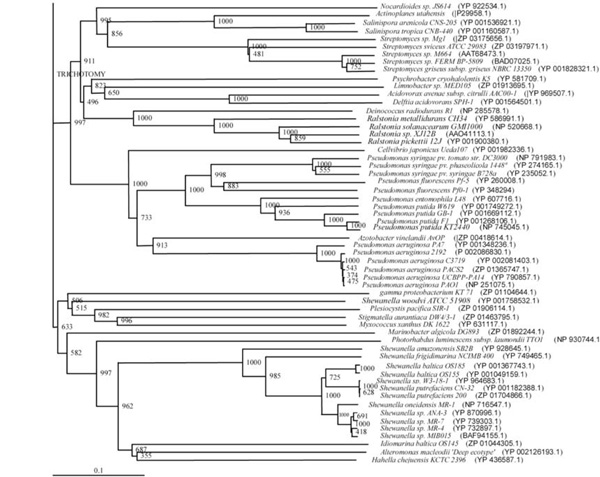
Phylogenetic tree (Lower half) based on protein sequences of organisms containing conserved domains with respect to Ralstonia sp. XJ12B AHL acylase as reference (For Taxonomic details refer to Additional file 3). A Neighbor-Joining analysis with Jukes-Cantor correction and bootstrap support was performed on the protein sequences. Bootstrap values are given at nodes, 1000 bootstrap replicates were run. The leaf contains Accession number and name of the organism.
SELECTION OF SEQUENCES BY HOMOLOGY SEARCH
The number of sequences in the each Table was reduced with respect to the homology among the same species using multiple sequence alignment - Neighbor Joining algorithm. We selected the sequence if the value of divergence was more than 10% [38], others could be deleted. The homology search helped to reduce the number of sequences in the each Table to approximately 100. This was done primarily to bring down redundancy and enable preparation of phylogenetic trees of reasonable size.
MULTIPLE SEQUENCE ALIGNMENT (MSA) AND RECONSTRUCTION OF PHYLOGENETIC TREE
For the reconstruction of the tree we have collected all the protein sequences for AHL-lactonase and AHL-acylase from NCBI, for each Table nearly 100 sequences were selected. ClustalX 2.0.10 [39] and Tree view 1.40 [40] were used for the multiple sequence alignments and phylogenetic tree reconstruction. Neighbor Joining algorithm was used for the tree reconstruction and Boot Strap value used was 1000 (default value). And the tree constructed was visualized with the Tree view software. Multiple aligned sequences were viewed using Jalview to find out the conserved region among them [41, 24].
SIGNATURE ANALYSIS & FINDING REGULAR EXPRESSION, USING MEME
MEME (Multiple EM for Motif Elicitation) is used for searching for novel motifs or signatures in sets of biological sequences. MEME works by searching for repeated, ungapped sequence patterns that occur in the DNA or protein sequences [43-45]. MEME searches can be performed via the web server (http://meme.nbcr.net) and its mirror sites [44]. The same web server also allows access to motif alignment and search tool to search sequence databases for matches to motifs. To successfully discover motifs with MEME, it is necessary to choose and prepare the input sequences carefully. Ideally, the sequences should be <1000 base pairs long [46].
Data sets in FASTA format were submitted group wise in MEME program Version 4.0.0 (http://meme.nbcr.net/meme4/cgi-bin/meme.cgi). In order to obtain maximum number of motifs in our sequences, we modified default settings from 3 motifs to 10 motifs. MEME search stops when this number of motifs has been found, or when none can be found with E-value less than 10000 (http://meme.nbcr.net/meme4/meme-input.html#width). We used default setting zero or one motif per sequence to get the occurrence of single motif which is distributed among the sequences. The default value of motif widths, set between 6 (minimum) and 50 (maximum) were modified and re-set between 25 and 30, respectively. Each of the 10 signatures (25 to 30 nucleotides long) (Table S2 to S17) was checked for its frequency of occurrence among all the sequences of a particular taxonomic group and the ones with highest frequency and did not appear in other taxonomic groups were considered unique to a particular reference taxonomic group.
In our analysis, sequences from each taxonomic group were analyzed for signature using MEME [43]. The MEME result gives a graphic representation and Motifs in regular expression format. MEME analysis were conducted for each taxa like Actinobacteria, Firmicutes, α-, β-, γ-, and δ-Proteobacteria, Ascomycota, Bacteroidetes, Euryarchaeota. The number of sequences of γ-Proteobacteria with reference to acylase from Ralstonia sp. Table were too large to be accommodated on line by the MEME Software (Word limit up to 60,000). In this case, we segregated the members of γ-bacterial group and made three case studies and searched for the signatures in i) Pseudomonas spp., ii) Shewanella spp. and iii) rest of the organisms (Designated as Others). The signatures obtained from the MEME program were cross-checked in other Tables and the other taxa in the same Table using online tool Sequence Manipulation Suite SMS [47].
VALIDATION OF SIGNATURES AND REGULAR EXPRESSIONS
The unique signatures - obtained after crosschecking with other taxonomic groups - were used as query sequence to BLAST against the sequenced microbial genomes available in NCBI database (http://www.ncbi.nlm.nih.gov/), to validate them [45]. Phylogenetic trees based on complete sequence of the gene and the strings based on the signatures were compared for AHL-lactonase among Firmicutes and AHL-acylase among γ-Proteobacteria – Pseudomonas spp. For developing strings, each of the 10 signatures was represented by a single alphabet - A to J.
RESULTS
Quorum sensing phenomenon has been reported from a wide range of organisms, which employ these primarily to sense environmental stress by releasing signal molecules: i) AHL by Gram-negative bacteria and ii) oligopeptides by Gram-positive bacteria. In contrast, quite a few organisms also have the abilities to degrade these signal molecules to ensure their survival. Here we are presenting the phylogenetic diversity of AHL inactivating enzymes: AHL-lactonases and AHL-acylases, which act by hydrolyzing the lactone ring and by cleaving the acyl chain, respectively.
PHYLOGENY OF AHL-LACTONASE AND AHL-ACYLASE
Using Bacillus sp. SB4 AHL-lactonase (Accession No. AAR854821) (Fig. 1) amino acid sequences as reference, BLAST resulted in detecting 110 sequences in different organisms showing completely conserved domains (Table S1). Sequences for this enzyme were found to be distributed among organisms belonging to Actinobacteria, Acidobacteria, Bacteroidetes, Chloroflexi, Deinococcus-Thermus, Firmicutes, α-Proteobacteria, β-Proteobacteria, γ-Proteobacteria, δ-Proteobacteria, Euryarchaeota, Crenarchaeota, Sphingo-bacteria, Spirochaetales, Nitrospirales, and Planctomycetes. Among these, two major groups of organisms possessing AHL-lactonase were represented by Firmicutes – 12 genera and 22 spp. and α-Proteobacteria – 18 genera and 22 spp. (Table S1).
On the other hand, with Ralstonia sp. XJ12B AHL-acylase (Accession No: AAO41113.1) (Fig. 1) as reference, BLAST resulted in detecting 94 sequences of different organisms showing completely conserved domains (Table S1). The distribution of sequences for AHL-acylase was rather limited in comparison to AHL-lactonase. It was found among Actinobacteria, Cyanobacteria, Bacteroidetes, Deinococcus-Thermus, Firmicutes, α-Proteobacteria, β-Proteo-bacteria, γ-Proteobacteria, δ-Proteobacteria, Euryarchaeota and Crenarchaeota. The distribution pattern of this enzyme was in clear contrast to that of AHL-lactonase. The largest group of organisms (59) possessing AHL-acylase belonged to γ-Proteobacteria - 20 genera and 31 spp. Here, Firmicutes and α-Proteobacteria were among the smaller groups constituted by 3 and 10 genera, respectively. The frequency of occurrence of AHL-acylase was relatively low among other Proteobacteria. Another contrasting scenario was observed among cyanobacterial members, which were found to possess only AHL-acylase. On the other hand, organisms belonging to Acidobacteria, Sphingobacteria, Spirochaetales, Nitrospirales and Planctomycetes were found to possess only AHL-lactonase (Table S1).
Multiple sequence alignment of all the sequences revealed that conserved regions for gene encoding for AHL-lactonase and AHL-acylase are located at the positions ranging i) from 467 to 484, 557 to 571 and 737 to 755 and ii) from 236 to 254 and 1892 to 1907 nts, respectively (Table 1). These regions can be used for designing ‘universal’ primers for amplifying genes from diverse taxonomic classes. In addition, certain conserved regions were also observed within each taxonomic group, which can be exploited for amplifying genes specific to them. The following conserved sequences can be used for amplifying lactonase: i) Firmicutes - TA[CT]ATTA at positions 535 to 542 nts, and ATATCGT at 774 to 780 nts, ii) β-Proteobacteria - CGCAGACCA at 681 to 689 nts, and CGG[AC]CG at 521 to 523 nts, etc. Similar regions were also observed among γ-Proteobacteria, δ-Proteobacteria and Ascomycota for lactonase gene (Table 1) and acylase type gene among Actino-bacteria, Cyanobacteria, α-Proteobacteria, β-Proteobacteria and Bacteroidetes (Table 1).
Location of Conserved Sequences in AHL-Lactonase and AHL-Acylase Genes of Taxonomically Diverse Bacterial Groups
| Taxonomic Group | Position (nts)a | Conserved Regions |
|---|---|---|
| AHL-lactonase gene | ||
| Allb | 467-484 | TGGTC[AG]AAATGCAGATGGGT |
| 557-571 | ATTTCGACCATGCCG | |
| 737-755 | AGCGATCACGCGTGTCC | |
| Actinobacteria | 560-570 | TCGACCACG[CG]CG |
| Firmicutes | 535-542 | TA[CT]ATTA |
| 559-566 | TTCGATCA | |
| 774-780 | ATATCGT | |
| Alphaproteobacteria | 562-571 | GACCAT[ACG]TCGT |
| Betaproteobacteria | 521-523 | CGG[AC]CG |
| 681-689 | CGCAGACCA | |
| Gammaproteobacteria | 527-534 | ATGTCGAG |
| 548-552 | CCCAT | |
| Deltaproteobacteria | 518-522 | A[GT]CCC |
| Euryarchaeota | 560-569 | T[CT]GACCACGC |
| Ascomycota | 338-347 | TTGAXCATCC |
| 520-539 | ATCAAAGACGTCAAGGCAGT | |
| 550-571 | CA[CT]TTACATCTCGA[CT]CACGCAG | |
| 628-633 | CACGAG | |
| AHL-acylase gene | ||
| Allc | 236-254 | TGATCGCCGAAATCCGGCG |
| 1892-1907 | GTATGGGTCCAGGCCA | |
| Actinobacteria | 704-713 | TACCGGGTCG |
| 1177-1181 | AC[AC]C[CG]GTCA[CG]C | |
| 1208-1217 | CCA[CG]GAGCTG | |
| Cyanobacteria | 1522-1527 | TTTGAAA |
| Alphaproteobacteria | 486-495 | TCGATGCG |
| Betaproteobacteria | 328-340 | GCGCAGGCAACC |
| Gammaproteobacteria | 309-320 | CGGCATCGGTTA |
| 586-595 | GGCTTCAACC | |
| Bacteroidetes | 1490-1497 | TC[CG]GCATA |
a: Nucleotides in the gene from 5’ to 3’.
b: Gene sequences from taxonomic groups listed under AHL-lactonase gene.
c: Gene sequences from taxonomic groups listed under AHL-acylase gene.
Potential Cases of Horizontal Gene Transfer of Acyl-Homoserine Lactone-Acylase and –Lactonase
| Incongruent Phylogenetic Associations | Boot Strap Value | |
|---|---|---|
| Organism(s) | Taxonomy | |
| Bacillus sp. SB4 (AHL-lactonase)a | ||
| Moorella thermoacetica ATCC 39073 | Firmicutes | 1000 |
| Burkholderia graminis C4D1M | β-Proteobacteria | |
| Mycobacterium sp. MCS | Actinobacteria | 820 |
| Rubrobacter xylanophilus DSM9941 | ||
| Granulibacter bethesdensis CGDNIH1 | α-Proteobacteria | |
| Acidiphilum cryptum Jf-5 | ||
| Agrobacterium tumefaciens | ||
| Deinococcus radiodurans R1 | Deinococcus-Thermus | 1000 |
| Xylella fastidiosa | γ-Proteobacteria | |
| Ralstonia sp. XJ12B ( AHL-acylase)a | ||
| Solibacter usitatus Ellin 6076 | Acidobacteria | 1000 |
| Plesiocystis pacifica SIR-1 | δ-Proteobacteria | |
| Deinococcus radiodurans R1 | Deinococcus-Thermus | 1000 |
| Ralstonia metallidurans CH34 | β-Proteobacteria | |
| R. solanacearum GM1000 | ||
| Ralstonia sp. XJ12B | ||
a: Reference organism.
Taxonomic Distribution of Organisms (Genera) Possessing Enzymes Acyl-Homoserine Lactone-Acylase as well as – Lactonase
| AHL-Lactonase | AHL-Acylase | ||
|---|---|---|---|
|
Bacillus sp. SB4a (GenBank AAR85482.1) |
Motifb |
Ralstonia sp. XJ12Ba (GenBank AAO41113.1) |
Motifc |
| Actinobacteria | |||
| Streptomyces clavuligerus ATCC 27064 | + | Streptomyces sp. FERM BP-5809 | + (I54-D61) |
| Streptomyces sp. M664 | + (I54-D61) | ||
| Streptomyces sp. Mg1 | + (I54-D61) | ||
| Streptomyces griseus subsp. griseus NBRC 13350 | + (I54-D61) | ||
| S. sviceus ATCC 29083 | + (L54-D61) | ||
| Deinococcus-Thermus | |||
| Deinococcus radiodurans R1 | + | Deinococcus radiodurans R1 | + (I58-S65) |
| D. geothermalis DSM 11300 | + | ||
| Alphaproteobacteria | |||
| Hyphomonas neptunium ATCC 15444 | + | Hyphomonas neptunium ATCC 15444 | + (I55-S62) |
| Betaproteobacteria | |||
| Ralstonia eutropha JMP134 | + | Ralstonia sp. XJ12B | + (I49-D56), + (I52-S59) |
| Ralstonia eutropha H16 | + (I62-D69) | ||
| R. metallidurans CH34 | + (I51-S58) | ||
| R. pickettii 12J | + (I51-D58) | ||
| R. solanacearum GMI1000 | + (I50-D57) | ||
| Gammaproteobacteria | |||
| Photorhabdus luminescens subsp. laumondii TTO1 | + | Photorhabdus luminescens subsp. laumondii TTO1 | + (I57-S64) |
| marine gamma proteobacterium HTCC2080 | + | marine Gamma proteobacterium HTCC2143 | + (I43-D53) |
| marine Gamma proteobacterium HTCC2207 | + (L106-D113) | ||
a: Reference organisms
b: Motif for AHL-lactonase: His106-X-Asp108-His109-59X-His169-21X-Asp191
c: Motif for AHL-acylase: amino acid – Ile (I) /Leu (L)50 and Ser(S) /Asp (D)57
Characteristics of Unique Nucleotide Signatures for Acyl-Homoserine Lactone-Acylase and –Lactonase Showing Closest Match to the Respective Taxonomic Group
| Taxonomic Group | Signature | Source Organism | |
|---|---|---|---|
| Nucleotide Sequence | No. | ||
| AHL-lactonase with reference to Bacillus sp. SB4 | |||
| Actinobacteria | CGTGAGCTCGAGTACGGCCTGTCGAATCACa | M8 | Mycobacterium sp.MCS [gi|108796981:1991857-1992693] |
| Firmicutes | TCACTTACATTTTGATCATGCAGGAGGAAA | M1 | Bacillus sp. SB4 [gi|40388446|gb|AY483161.1|] |
| TCCGGTTCAGTTTTATTAACGATTGATGCA | M10 | ||
| Alphaproteobacteria | GTTGTGCAGTCGCACCTGCATCTCGATCATa | M1 | Agrobacterium tumefaciens [gi|17223781:3565-4356] |
| ATCCGCAAGGACTTCGACAAGCCGGGCCTa | M4 | ||
| Betaproteobacteria | GTGGATACGGAAGACGGCGTGGTGCTGATa | M7 | Burkholderia graminis [gi|170694799:130043-130822] |
| Gammaproteobacteria | AATCCGGTATCAATCAGTACAAGGCCTCGAa | M2 | Acinetobacter baumannii AYE [gi|169794206:627842-628654] |
| Deltaproteobacteria | ATGGTCGTTATGCAGGTGCGTGTGTATGACa | M1 | Desulfovibrio desulfuricans G20 [gi|78355047:1350539- 1351285] |
| Ascomycota | TACACGAGGAAGAGTTCAAGCATGCATGCTa | M2 | Aspergillus oryzae RIB40 [gi|169784499|ref| XM_001826659.1|] |
| GATCAATTCCATGTGCGGGAGAACTACGAGa | M3 | ||
| Euryarchaeota | All signaturesa | ||
| AHL-acylase with reference to Ralstonia sp. XJ12B | |||
| Actinobacteria | CGGGTTCGCCGCCGGGTACAACGCGTGGATa | M2 | Streptomyces sp. M664 [gi|49781326:185-2599] and Streptomyces sp. FERM BP-5809 [gi|40786372:948-3362] |
| GTCATCCGGTACACGGAGTACGGCATTCCGa | M3 | ||
| Bacteroidetes | ATCGAGATGCTTGAAGGGGATTCCTCGAa | M3 | Salinibacter ruber DSM 13855 [gi|83814055:1438503-1440524-] |
| GGCTTCGGGGGGAGATGGTCTTTTTGa | M7 | ||
| TTTGACGAGATGGAGGGCGGGa | M8 | ||
| Cyanobacteria | GCTGGTTCCAATGCTTGGGCGATCGCACCTa | M5 | Cyanothece sp.[gi|126661348:2396-4477-] |
| Alphaproteobacteria | ATGACTCCGGCAAGCCCGAAGAAGAAAGGCa | M7 | Erythrobacter litoralis HTCC2594 [gi|85372828:319908-322139] |
| GATATTGCCCTCGGCATCGCCATAGATGAAa | M10 | ||
| Betaproteobacteria | CCAGCCGCATGCGCAGGGTTTCCTGGGTCCa | M3 | Ralstonia eutropha H16 [gi|113866031:2083444-2085906] |
| TCGGCACGCGCAGGTTGAACAGCGGCGACAa | M7 | ||
| Gammaproteobacteria: Pseudomonas spp. | CTGCCGGTGGTCAACATCGGCTTCAGCCGC | M1 | Pseudomonas aeruginosa PA7 [gi|152983466:2955521-2957803] |
| TTCGTACAGAACTCCAACGACAGCGCCTGG | M2 | ||
| GATGCGTTTCTACCAGATGCACCTGACCAT | M3 | ||
| ATGCTGCTGGCCAACCCGCATTTCCCCTGG | M4 | ||
| CGGCATCGGCTACGCCTACGCGCGCGACA | M6 | ||
| GCGCGATGCCAACCTGGAGAACACCCGGGT | M7 | ||
| CCGCTGGACCGCCTATGGCGTGCCGCACAT | M9 | ||
| ATCCCCTGGGTCAACACCCTGGCCGCGGA | M10 | ||
| Gammaproteobacteria: Shewanella spp. | CAGATAATAAAGGCAACGCCTTCTACATCGa | M1 | Shewanella woodyi ATCC 51908 [gi|170724370:166858-169239] |
| CCCTTTGAGCGTGCCCCCAAAGTGATGCGTa | M2 | ||
| GGCCGTGATAAAACCGAATCTGGTAGCGGTa | M3 | ||
| TTAATCGGCTTCAATGAACATGTTGCTTGGa | M6 | ||
| GGTTATGGGCAAGCCTATGCCCATGCTCAGa | M10 | ||
| Others Gammaproteobacterial members | TTATCCGCTTCGATGTGCGGCGCGCCGAAa | M8 | Hahella chejuensis KCTC 2396 [gi|83642913:5633846-5636326] |
a: Sequence homology searches using BLAST for these signatures showed best hits for the query sequence but the frequency among the top 50 hits was quite low.
PHYLOGENETIC DISCREPANCIES
The phylogenetic trees (Figs. 2-5) based on organisms found to possess AHL-lactonase and AHL-acylase followed the taxonomic distribution, which is expected on the basis of their 16S rDNA sequences. A few phylogenetic discrepancies were observed for AHL-lactonase (Figs. 2,3, Table 2), the notable being the associations between i) Moorella thermoacetica ATCC 39073 (Firmicutes) and Burkholderia graminis C4D1M (β-Proteobacteria) (Boot Strap Value, BV 1000), ii) Actinobacteria (Mycobacterium sp. MCS and Rubrobacter xylanophilus DSM9941) and members of α-Proteobacteria (Granulibacter bethesdensis CGDNIH1, Acidiphilum cryptum Jf-5 and Agrobacterium tumefaciens) (BV 820), and iii) Deinococcus radiodurans R1 (Deinococcus-Thermus) and Xylella fastidiosa (γ-Proteobacteria) (BV 1000). Similarly, certain taxonomic discrepancies were also recorded in the phylogenetic tree (Figs. 4,5, Table 2) drawn with respect to sequences of organisms possessing AHL-acylase: i) Solibacter usitatus Ellin 6076 (Acidobacteria) and Plesiocystis pacifica SIR-1 (δ-Proteobacteria) (BV 1000), ii) D. radiodurans R1 (Deinococcus-Thermus) and Ralstonia metallidurans CH34, R. solanacearum GMI1000 and Ralstonia sp. XJ12B (β-Proteobacteria (BV 1000). Since these associations between widely distributed organisms show very high BVs, these may be interesting cases of horizontal gene transfers.
ORGANISMS POSSESSING GENES FOR AHL-LACTONASE AND AHL-ACYLASE
Sixteen bacterial strains of 5 different genera and a group of 3 strains belonging to marine gamma proteobacterium were found to possess genes for both the enzyme types: AHL-lactonase and AHL-acylase (Table 3). The diversity of these 19 strains was evident by their distribution among 5 different taxonomic groups: i) Streptomyces spp. (Actinobacteria), ii) Deinococcus spp. (Deinococcus-Thermus), iii) Hyphomonas sp. (α-Proteobacteria), iv) Ralstonia spp. (β-Proteobacteria), v) Photorhabdus sp. (γ-Proteobacteria), vi) marine gamma proteobacterium. Three out of these 19 strains can be more clearly classified as those which possessed genes for both the enzyme types: i) D. radiodurans R1, ii) Hyphomonas neptunium ATCC 15444 and iii) Photorhabdus luminescens subsp. laumondii TTO1. This observation is supported by the presence motifs for lactonase and acylase in these strains.
SIGNATURE ANALYSIS – FREQUENCY AND DISTRIBUTION PATTERN
Ten signatures (nucleotides) were obtained through MEME for different taxonomic groups with reference to: i) AHL-lactonase gene (aiiA) of Bacillus sp. SB4 Accession No. AAR85482.1 (Figs. S1 to S8 and Tables S2 to S9) and ii) AHL-acylase gene (aiiD) of Ralstonia sp. XJ12B Accession No. AAO41113.1 (Figs. S9 to S19 and Tables S10 to S17). (See Additional File 4 for a description of data in Tables S2 to S17 and Figs. S1 to S17). Nucleotide signatures found to be present at high frequency within the taxonomic group and absent from all other groups were initially classified as unique. The BLAST (against the NCBI database) result validated the uniqueness of these signatures (Table 4). A high correlation was recorded for signatures M1 and M10 of Bacillus sp. SB4 and their occurrence among a large number of organisms possessing AHL-lactonase (BLAST hits with high frequency). Similarly, among the different signatures identified within each taxonomic group for AHL-acylase, those retrieved from Pseudomonas spp. within γ-Proteobacteria gave the BLAST hits with high frequency for organisms of this taxonomic group. Sequence homology searches using BLAST for all other signatures showed best hits for the query sequence from the respective taxonomic group but their frequency among the top 50 hits was quite low.
DISCUSSION
Bacteria with Ability to Quench QS Signals
With the information available in the literature on the presence of AHL-lactonase and AHL-acylase activities in R. erythropolis W2 [30-32], we also looked for certain other organisms with potential to produce both these enzyme types. Our in silico study allowed us to identify organisms from five different genera with potential to produce both types of AHL-degrading enzymes. The three strains i) D. radiodurans R1, ii) H. neptunium ATCC 15444 and iii) P. luminescens subsp. laumondii TTO1 are among the top contenders for producing both the enzzyme type from an individual strain. However, more such strains can be tracked among strains belonging to Streptomyces spp. (Actinobacteria), ii) Ralstonia spp. (β-Proteobacteria), and marine gamma proteobacterium. In order to reduce the efforts needed to search organims, conserved regions can be used for designing primers for amplifying these genes. Signature sequences deduced in this study can be used as an additional tool to assign these sequences to respective taxonomic groups, at least in some cases.
Another interesting features of this in silico study is that R. solanacearum GMI1000 (β-Proteobacteria) has been found to possses gene and motif for AHL-acylase. This observation finds support from the work which demonstrates the presence of AHL-acylase in R. solanacearum GMI1000, through cloning and expression of aac gene [25]. It led to inactivation of four different AHLs [25]. Another interesting observation was the presence of genes for quenching the QS signals in archaeal member A. fulgidus. Although archaea have not been reported so far to be directly involved in causing infectious diseases [48], however, there are evidences of their presence in endodonic infections [49]. The presence of these genes support the possibility of their role in infections.
MOTIFS OF AHL-LACTONASES AND AHL-ACYLASES
AHL-lactonase is a member of metallo-hydrolase superfamily containing the motif: His104-X-His106-X-Asp108-His109, which resembles zinc binding motif of several other metallo-enzymes such as glyoxalase II, aryl sulfatase and β-lactamase. The motif His106-X-Asp108-His109-59X-His169-21X-Asp191 has been reported to be essential for AHL-lactonase activity [18, 22].
A very interesting scenario was observed while searching for motifs essential for AHL-lactonase enzyme activities. It was observed to be that motif essential for AHL-lactonases - His106-X-Asp108-His109-59X-His169-21X-Asp191 was present in all the organisms having potential for AHL-lactonase except R. solanacearum GMI1000, A. baumannii AB0057, A. baumannii ACICU, A. baumannii AYE, Acinetobacter sp. ADP1 and Pseudomonas stutzeri A1501 (Table 3). Incidentally, these latter 6 bacteria lacked even the motif specific for metallo-enzymes in genral- His104-X-His106-X-Asp108-His109. Among the organisms found to possess both the motifs – all belonged to Bacillus spp.: B. anthracis str. Ames, B. cereus, B. cereus AH1134, B. cereus AH820, B. cereus ATCC 10987, B. cereus ATCC 14579, B. cereus B4264, B. cereus E33L, B. cereus G9241, B. cereus NVH0597-99, Bacillus sp. 240B1, Bacillus sp. 42, Bacillus sp. 91, Bacillus sp. A24, Bacillus sp. COT1, Bacillus sp. CSX-1, Bacillus sp. SB4, B. subtilis, B. thuringiensis, B. thuringiensis serovar alesti, B. thuringiensis serovar canadensis, B. thuringiensis serovar galleriae, B. thuringiensis serovar israelensis ATCC 35646, B. thuringiensis serovar jinhongiensis, B. thuringiensis serovar kim, B. thuringiensis serovar konkukian str. 97-27, B. thuringiensis serovar kurstaki, B. thuringiensis serovar kyushuensis, B. thuringiensis serovar ostriniae, B. thuringiensis serovar oswaldocruzi, B. thuringiensis serovar pakistani, B. thuringiensis serovar toumanoffi and B. weihenstephanensis KBAB4.
Available literature reveals that AHL-acylases are structurally similar with respect to the two amino-acids at positions 50 and 57 [18] as a) AiiD of Ralstonia sp. XJ12B has Ile50 and Ser57 [26], b) PvdQ of P. aeruginosa PAO1 has Leu50 and Asp57 [27], and c) AhlM of Streptomyces sp. has Leu50 and Ser 57 [50]. These share features with Ntn hydrolases including the signal peptide, α- and β-subunits [51].
On the other hand, Cephalosporin-acylase has Gln and Arg at positions 50 and 57 respectively [18]. Our analysis reveals that most of the organisms had amino-acids of AHL acylase in a slightly different positions than those reported in the literature (Table 3). Unique features were observed in i) Streptomyces spp., where the amino acids of motif for acylase resembled either those found in PvdQ or were a combination of Ile of AiiD and Asp of PvdQ, ii) Ralstonia spp., where Ser was substituted with Asp and also that Ralstonia sp. XJ12B was found to posses two motifs – a) Ile52 and Ser59 and b) Ile49 and Asp56. In view of the fact that in spite of such a large number of organisms, which had the conserved domains for AHL-acylase, only few seem to have retained the property observed at respective positions as reported in literature [18, 26, 27, 50]. It may not be too inappropriate to conclude that amino-acids at positions 50 and 57 might have changed during the course of evolution.
A few other organisms which deserve attention are: i) Burkholderia spp. (β-Proteobacteria), which had motifs for lactonase and cephalosporin acylase, ii) Pseudomonas sp. (γ-Proteobacteria), which possessed two motifs for AHL-acylase and one for cephalosporin acylase, and iii) A. baumannii AYE (γ-Proteobacteria), which showed motifs for cephalosporin acylase.
USING SIGNATURES TO DESIGN PRIMERS
Bacillus spp. and Pseudomonas spp. have been reported to possess repeating elements in the 16S rDNA [2, 45]. We observed such repeats in the AHL-lactonase and -acylase gene in different members of these two genera. Individually, these signatures were validated by using them as query sequences in BLAST search. Since the frequency of repeating elements in this gene was quite high in the case of Pseudomonas spp., we also checked them for regular expressions [52]. We observed a very good co-relation between the full length gene sequence and the strings (Fig. S18a, b). The nucleotide signatures identified in our study for each taxonomic group may enable one to design primers to amplify genes for AHL-lactonase [20, 24, 53] or AHL-acylase and identify a possible taxonomic group within a microbial community.
THE ROLE OF QUORUM QUENCHERS
The role of QQ enzymes in vivo is debatable [54]. They may be involved in metabolic pathways where AHL and its products may be used as energy and carbon sources -such as by Variovorax paradoxus and Arthrobacter spp. [55-57]. AHL-acylase PvdQ from P. aeruginosa is thought to be involved in biosynthesis of protein (pyoverdine) necessary for acquisition of iron [28, 54, 58]. The role of the enzymes – lactonase and acylase for QQ has been proposed to prevent food, prevent biofilm growth, surface fouling, treatment of bacterial infections, etc. [59]. It can act as a potential drug, which will selectively block the virulence and pathogenic traits [60]. Heterologous expression of Bacillus AiiA lactonase in Burkholderia thailandensis [24], in P. aeruginosa PAO1 [23], in Vibrio harveyi [53], in E. carotovora [22] have been found to disrupt QS mediated properties. In fact, certain antibiotics become effective on biofilms which have been exposed to AHL degrading enzymes [61], which imply a combination of the two treatments can be complementary and effective in controlling the pathogenic organism(s). Although there may be barriers to their diffusion within the microbial communities in the soil environment, however, their usage as a drug may not face similar barriers and may prove effective.
CONCLUSION
Using available metabolic and genomic databases databases, we have been able to trace potential organisms such as i) Deinococcus radiodurans R1, ii) Hyphomonas neptunium ATCC 15444 and iii) Photorhabdus luminescens subsp. laumondii TTO1 to possess genes for AHL-lactonases and –acylases. In addition, all the cyanobacterial members were found to possess genes only for AHL-acylase whereas members of Acidobacteria, Sphingobacteria, Spirochaetales, Nitrospirales and Planctomycetes were found to possess genes only for AHL-lactonase. It also revealed those bacteria which evolved to produce either of the two enzymes, an indication of the environmental stresses encountered during their evolution. The presence of genes for quenching the QS signals in archaea has further increased the range of organisms which can be exploited for producing these enzymes for preventing food spoilage, bioremediation and design drugs against bacterial infections.
Phylogenetic analyses and multiple sequence alignment of the gene sequences coding for the two enzymes revealed consensus sequences, which can be used to design primers for amplifying these genes even among mixed cultures and metagenomes. Nucleotide signature sequences deduced in this study can be used as an additional tool to assign these sequences to respective taxonomic groups. A few phylogenetic discrepancies observed for AHL-lactonase and –acylase may imply HGT and help us identify those organisms which can be subjected to genetic engineering with a relative ease.
It has been proposed that since lactonase activity is not influenced by the acyl chain length of the AHL signal molecule, hydrolysis of the ring may be more effectve approach for QQ. As a wide range of Bacillus spp. have an ability to produce lactonase, it may be the organism of choice in future. The usage of Bacillus is quite beneficial since it has been labeled as industrial workhorse and accorded the designation GRAS (Generally Regarded As Safe) by FDA. These organisms can be exploited for producing drugs against pathogens.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers web site along with the published article.
ACKNOWLEDGEMENTS
We are thankful to Directors of Institute of Genomics and Integrative Biology, CSIR; National Environmental Engineering Research Institute, CSIR grant SIP 16 for providing necessary funds, facilities and moral support.
AUTHOR’S CONTRIBUTIONS
VCK has contributed towards the conceived and designed the experiments, analyzed the data and wrote the paper.
CRS and HJP have performed the experiments, and contributed reagents/ materials/ analysis tools. All authors read and approved the final manuscript.

