All published articles of this journal are available on ScienceDirect.

Optimising Bacterial DNA Extraction from Faecal Samples: Comparison of Three Methods
Authors Info & Affiliations
Abstract
Culture independent methods are used widely in diagnostic laboratories for infectious disease Isolation of genomic DNA from clinical samples is the first and important step in the procedure. Several procedures for extracting DNA from faecal samples have been described, including different mechanical cell disruptors. To our knowledge, the use of TissueLyser as a mechanical disruptor on faecal samples before DNA extraction has not been previously described. The purpose of the study was to implement a method for preparing faecal samples for optimal DNA extraction. Thus, three different procedures for extracting DNA from human faeces were compared. This was done either by using the mechanical disrupter by Mini BeadBeater 8, or the TissueLyser both followed by DNA purification using QIAamp DNA stool MiniKit, in comparison with DNA extractions using QIAamp DNA stool MiniKit without any prior mechanical disruption, according to manufacturer’s instructions. The obtained DNA from the three procedures was analysed by DGGE, and the number of bands was compared between each procedure. There was no significant difference between the numbers of bacterial bands obtained from DGGE when using a TissueLyser or Mini BeadBeater 8, so the two different mechanical cell disruptors can be used comparably when isolating bacterial DNA from faecal samples. The QIAamp DNA stool MiniKit alone resulted in a reduced number of bands compared to the two mechanical disruption methods.
INTRODUCTION
Faecal samples contain a large variety of bacteria [1-3]. Using conventional culture techniques, only a small percentage of the bacterial flora is identified [2, 4-5]. With the introduction and implementation of molecular techniques in clinical microbiological diagnostic laboratories, the identification of bacteria present in faecal samples has increased dramatically [5-7]. The isolation of genomic DNA from clinical samples is the first step in studies of microbial diversity using cultivation independent methods. It is, therefore, important to obtain genomic DNA that is representative of the microbial communities present in the samples [8]. However, the use of molecular methods has introduced new problems, such as obtaining sufficient amounts of DNA and of high quality of intact DNA suitable for further processing i.e. PCR, from faecal samples [8-10]. Several procedures for extracting DNA from faecal samples have been described, including different mechanical cell disruptors [2, 11, 12]. None of the described methods have been compared systematically and in the same study using the same samples.
In this study, three methods were compared to evaluate the amount and quality of bacterial DNA extracted from human faecal samples. QIAamp DNA stool MiniKit without any prior sample disruption treatment was compared to two mechanical disruption sample preparations, i.e. the Mini BeadBeater 8 and the TissueLyser (Qiagen) used for animal tissue and plant disruption. Both homogenisations were followed by the QIAamp DNA stool MiniKit in order to compare bacterial DNA extractions.
MATERIALS AND METHODS
Human Faecal Samples
Fresh faecal samples were obtained from the Laboratory for Enteric Pathogens, Statens Serum Institut, Copenhagen, Denmark, submitted for routine culture. The subjects were between six months and four years of age, and all had diarrhoea; no other clinical information was obtained. Each sample was adjusted to 200 mg wet weight stool before storage at -80°C without glycerol, until processing 24 hours later.
Preparation of Samples
Each faecal sample was mixed with 1.4 ml ASL (from QIAamp DNA stool MiniKit) in a 2 ml tube and vortexed until the faecal sample was thoroughly homogenized. Samples for the two mechanical treatments were mixed with 0.2 g sterile zirconia/silica beads (diameter, 0.1 mm, Biospec Product, ROTH, Karlsruhe, Germany).
The mechanical disruption procedures were: the Mini BeadBeater 8 (BioSpec Products Inc., Bartlesville, Oklahoma, USA) and the TissueLyser system (Qiagen Retsch GmbH, Hannover, Germany). Both procedures were followed by DNA extraction using QIAamp DNA stool MiniKit, (QIAGEN, Hilden, Germany).
The Mini BeadBeater 8
Bead mill homogenization by Mini BeadBeater 8, with high speed agitation, was performed on faecal samples as described by [2]. The samples were homogenized for 4 min at 5000 rpm in a Mini BeadBeater 8.
TissueLyser
Bead mill homogenization with the TissueLyser system has not been described for faecal samples previously, therefore optimising the method was necessary.
The TissueLyser is a vibration apparatus providing high-throughput processing for effective disruption of biological samples including bacteria, animal soft tissue and plant tissue [13]. The TissueLyser provides disruption and homogenization achieved through the beating and grinding effect of the beads on the sample material as they shake together in the grinding vessels. Disruption for 4 min, 6 min and 8 min at 30 Hz were performed in an adapter set for 2 x 24 samples. To prevent variation in sample homogenization, the adaptor was removed from the TissueLyser and disassembled in a reverse order after the first disruption, to ensure a uniform sample homogenisation.
QIAamp DNA Stool MiniKit (Treated and Untreated Samples)
All samples were centrifuged for 2 minutes at 14,000 rpm prior to the DNA extraction. Extraction of DNA from the supernatant was performed following the protocol of QIAamp DNA stool Mini Kit (QIAGEN, Hilden, Germany) as described by the manufacturer with the following modifications: lysis temperature, 95°C [6]. DNA was measured spectrophotometrically (Ultrospec 1100 pro, Amershan Biosciences).
PCR Amplification for DGGE
The V3 region of the 16S-rRNA with a length of 236 base pairs was amplified with universal bacterial primers. The forward primer PRBA338, positions 338-357 (E. coli numbering) (5´ACTCCTACGGGAGGCAGCAG) [14] and reverse primer PRUN518, positions 518-534 (E. coli numbering) (5´ATTACCGCGGCTGCTGG) [4, 15]. The forward primer PRBA 338 primers were at the 5´ end labelled with a GC clamp (5´CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG) [4]. All primers were purchased from MWG-BIOTECH AG, Ebersberg, Germany. The final volume of each PCR mixture was 50 µl. The amplification reaction mixture was as follows: 1.25 U of Taq DNA polymerase (Promega, Madison, Wis), 5 µl 10x PCR buffer, [1.5 mM MgCl2], 1 µl of deoxynucleutide triphosphate at a concentration of 10mM, 0.5 µM of each primer, 1 µg of DNA template, 0.5 µl BSA (10 mg/ml). The samples were amplified with a Peltier Thermol Cycler, and the following conditions were used: preheating 94°C for 5 min; 30 cycles of denaturing 94°C for 45 sec, annealing 55°C for 30 sec., elongation 72°C for 45 sec.
DGGE of PCR Amplicons
The PCR fragments were separated by DGGE as described by Myuzer [4] with the DCode System as manufacturer prescribes (Bio-Rad Laboratories, Hercules, USA), 8% Polyacrylamide (vol/vol) (ratio of acrylamide:bisacrylamide [37.5:1]) in 0.5×TAE buffer; (pH 8.0) and using a gradient ranging from 30% to 65% denaturant (100% denaturant acrylamide corresponds to 7 M urea and 40% (vol/vol) formamide). The gels were cast using a gradient maker and a pump with a flow speed of 5 ml per min. After polymerization of the denaturing gel (2 hours), a 3% stacking gel without denaturing chemicals was cast, and an appropriate comb was subsequently inserted and left 10 min for polymerization. Gels were run at 60°C for 16 h at a constant voltage of 70 V in 0.5 × TAE buffer. After electrophoresis, gels were stained with SYBR-GOLD (Molecular Probes, Eugene, Org, USA) for 20 min, destained briefly in Milli-Q water and photographed on the Gel Doc system (Biorad).
RESULTS
DNA Amount Extracted from Faecal Samples
Faecal samples were divided into equal portions and treated by mechanical disruptors. Different time lengths of disruption were only tested for the TissueLyser, as indicated in materials and methods.
The highest amount of total DNA was obtained, when using the Mini BeadBeater 8 for four minutes and the TissueLyser for eight minutes (data not shown). Using the TissueLyser for more than six minutes showed partial degradation of the genomic DNA on agarose gel. Treatment with TissueLyser for 4 minutes did not yield the same amount of DNA compared to 4 minutes treatment with the Mini BeadBeater 8 (data not shown).
DNA for Further Processing
The quality of DNA was tested by performing PCR-DGGE that targeted all the bacteria present in the faecal sample. Similar results for the two mechanical methods for pre-treatment, i.e. the Mini BeadBeater for 4 min, and TissueLyser for 6 and 8 min were observed (Fig. 1). The QIAamp stool Minikit alone with no pre-treatment, resulted in a reduced number of bands on the DGGE. Furthermore, the TissueLyser treatment for 4 min was not sufficient (Table 1).
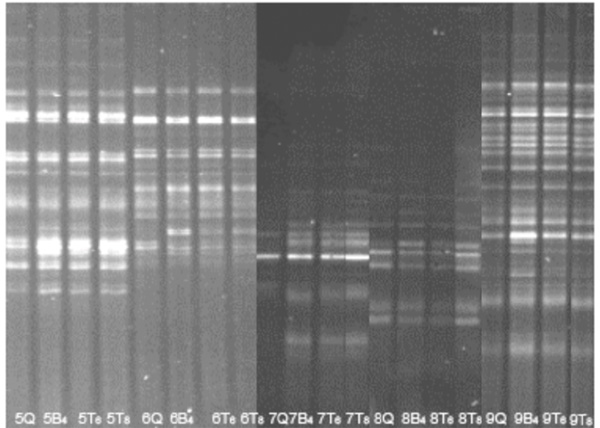
DGGE on DNA extracted by three different treatment methods from faecal samples. Example of DGGE runs of PCR products from five stool samples by different DNA extractions. DGGE profiles (30-65% denaturant) from faecal samples on DNA extracted by three different treatment methods and treatment times. (B) pre-treatment by Mini BeadBeater 8 (T) pre-treatment by TissueLyser, (Q) no pretreatment. All subsequent DNA extractions were performed by the QIAamp DNA stool MiniKit. The agitation time was 4 minutes for the Mini BeadBeater 8, and 6 minutes and 8 minutes for the TissueLyser. (The picture is compiled from two gel images, aligned according to the reference lane comprising an internal standard (not shown)).
DISCUSSION
Studies of intestinal microflora have conventionally been performed by bacterial culturing on selective media [16, 17]. Introduction of molecular techniques, particularly the use of PCR- DGGE, has revealed even more complex microbiological communities to exist in the intestine [2, 5, 18]. Several studies have presented different methods for DNA extraction, such as QIAamp DNA stool MiniKit, Mini Bead Beater 8 [2], and TissueLyser [13]. However, the described methods have not been compared systematically on the same samples and in the same laboratory setting. It is crucial in clinical microbiology to find a reproducible procedure for obtaining sufficient amounts of DNA, with no degradation products, from even a very small sample.
The results presented in this study show that a mechanical pre-treatment before using the QIAamp DNA stools MiniKit facilitates an efficient homogenizing and lysis, leading to the recovery of an increased amount of bacterial DNA (Table 1 and Fig. 1).
Number of DGGE Bands Obtained by PCR after Different Physical Disruption of the Samples
| DGGE bands | |||||
|---|---|---|---|---|---|
| Tissue Treatment | n | mean | Std dev | Min | Max |
| Qiagen only | 9 | 11 | 6 | 2 | 20 |
| Beadbeater 4 min | 9 | 12 | 7 | 3 | 26 |
| TissueLyser 4 min | 4 | 9 | 5 | 6 | 16 |
| TissueLyser 6 min | 5 | 15 | 7 | 10 | 25 |
| TissueLyser 8 min | 5 | 15 | 6 | 10 | 24 |
DGGE profiles (30-65% denaturant) from 9 faecal samples on DNA extracted by three different treatment methods. All subsequent DNA extractions were performed by the QIAamp DNA stool MiniKit.n: number of treated samples; mean: mean number of DGGE bands; Std dev: standard deviation of DGGE bands; min: minimum of DGGE bands; max: maximum of DGGE bands.
In this study, the TissueLyser showed similar results in disrupting genomic DNA from faecal samples compared to the Mini BeadBeater 8. We demonstrated that the DGGE bands obtained by treatment of 8 min with Mini BeadBeater were similar to those achieved when agitating 4 min and 6 min, although there was some level of DNA degradation in longer treatments (8 min). The two types of mechanical disruptions are both fast and with the same hands on time. The Mini BeadBeater 8 can handle 8 samples simultaneously, whereas the Tissuelyser handles 1-52 or 1-96 samples simultaneously. However, to prevent variation in sample homogenization, the adaptors have to be disassembled after the first disruption and the order reversed.
The study showed that the TissueLyser (6 min processing) is comparable with the Mini BeadBeater 8 (4 min processing) with regard to the quality and number of the DGGE bands (Table 1).
Furthermore, the mechanical disruption of the faecal samples before using the QIAamp DNA stool MiniKit improved the amount and quality of DNA and thereby the diversity in the bacterial bands in DGGE. We conclude that both mechanical devices can be used with comparable results, favouring the Mini BeadBeater 8 for a small number of samples, while the TissueLyser is recommended for processing of a large number of samples.
CONFLICTS OF INTERESTING
The authors have no conflicts of interest to report.
ACKNOWLEDGMENTS
Researcher Dr. Nan Li was partially financed by the Chinese Government.

