All published articles of this journal are available on ScienceDirect.

Strong Strand Composition Bias in the Genome of Ehrlichia canis Revealed by Multiple Methods
Abstract
Genes located on the two replicating strands are found to have two separate base/codon usages in Ehrlichia canis genome. Although strand-specific codon usage is not the first observation, for the first time we have applied multiple methods to the analysis of strand composition bias. By combining multiple methods, comprehensive and interesting results are obtained. Among three types of correspondence analysis (COA), absolute codon usages between genes on the two replicating strands are more distinct than relative synonymous codon usage (RSCU) and base usages are more sensitive than both types of codon usages. PR2-plots show that replication-induced bias is much higher than transcription/translation associated bias in the genome. By using the Z curve method, two common genomic characters, i.e., stronger strand composition bias and lower rearrangement frequency are found to exist in 11 obligate intracellular bacteria with separate base/codon usages. We hope more and more researchers will use the multiple methods to analyze strand composition bias in sequenced microbes, particularly obligate intracellular bacteria.
1.. INTRODUCTION
Chromosomal replication, particularly that for microbes, includes a set of asymmetric mechanisms, among which is a division into lagging and leading strands [1]. In 1991 strand-specific nucleotide composition bias was first found in genomes of echinoderm [2] and vertebrate mitochondria and then in several bacterial genomes [3]. With the rapid growth in the number of sequenced genomes, more and more bacteria are found to have the consistent strand composition bias [4]. That is to say, there is always the excess of bases G relative to C in the leading strands and of C to G in the lagging strands, which is frequently accompanied by the bias of T versus A. Now, the underlying causes of strand composition bias have not been completely understood [5]. Two published papers reviewed numerous explanations for the composition bias [6, 7]. These explanations either attribute the bias to replication induced mutation/repair asymmetry or to transcription/translation coupled mutation/repair asymmetry [5, 8]. For either kind of hypothesis, cytosine deamination of single-stranded DNA performs a vital role in the generation of strand composition bias [9].
In 1998 [10], an exceptionally strong strand composition bias was observed in Borrelia burgdorferi, the causative agent of Lyme disease. The bias is so strong that genes on the two replicating strands could be discriminated according to their codon usages. In the past decade, another 9 bacterial genomes were also found to have extremely strong composition bias [11-13]. In other words, genes on the two replicating strands were found to have separate base/codon usages in genomes of 10 bacteria. Interestingly, all of these bacteria are obligate intracellular [7]. Rocha attributed the association between obligate intracellular niches and strong strand composition bias to the extreme stability of most of these genomes [7]. He supposes that repeats induce frequent chromosomal rearrangements, and may thus reduce strand composition bias. On the other hand, Klasson and Andersson [14] found that strong strand composition bias in three endosymbionts Blochmannia floridanus, Blochmannia pennsylvanicus and Buchnera aphidicola coincided with loss of genes for replication restart pathways.
In almost all prior works that reported the separate codon usages of genes located on the two replicating strands, only one single method, namely correspondence analysis (COA) of relative synonymous codon usage (RSCU) was employed [15]. To deeply analyze and obtain more reliable, complete information about the phenomenon existing in special genomes, here we adopt three methods, i.e., COA, the Z curve and PR2-plot, to investigate strand composition bias in Ehrlichiacanis str. Jake genome. E. canis is an obligate intracellular member of the order Rickettsiales and infection with it could cause ehrlichiosis in dogs.
2.. MATERIALS AND METHODS
2.1.. The Database
The complete DNA sequence and annotation information of E. canis (AC number: NC_007354) were downloaded from GenBank ftp site (ftp://ftp.ncbi.nih.gov/genbank/). Totally, 925 protein-coding genes are listed in the annotation. Positions of replication origin and terminus the same with that provided by DoriC database http://tubic.tju.edu.cn/doric/. The origin is assigned to lie before gene Ecaj_0011, whereas the terminus between genes Ecaj_0446 and Ecaj_0447. Consequently, 532 genes are located on the leading strands and the other 393 on the lagging strands.
2.2.. Z Curve and Nine Variable of Phase-Specific Z Curve
The Z curve is a 3-D space curve constituting the unique representation of a given DNA sequence in the sense that for the curve and sequence each can be uniquely reconstructed from the other [16]. Denoting the cumulative occurring numbers of the bases A, C, G and T in a DNA sequence read from the 5' to the 3'-end by An, Cn, Gn and Tn, respectively, we define the Z curve in the following. The Z curve consists of a series of nodes Pn, where n = 1, 2, ..., N, whose coordinates are denoted by Xn, Yn and Zn. It is shown that [16]:
where A0 = C0 = G0 = T0 = 0 and hence x0 = y0 = z0 = 0. P0 is the origin of coordinate system. The connection of the nodes P0, P1, P2, ..., until PN one by one sequentially by straight lines is called the Z curve for the DNA sequences inspected.
As is known, a protein-coding gene has three codon positions. For a gene, we could make three Z curves for all codon positions and each of them has three components. Therefore, a gene could be represented by nine components. Each component curve could be fitted as a straight line by using least squares technique. To mathematically describe the gene, we use the slopes of their fitting lines to denote nine component curves. For simplicity, we often use mean frequencies of bases to replace fitting slope. The so generated nine values, denoted by u1-u9, are called as nine variables of phase-specific z curve. For details of this method, please refer [16]. In fact, the nine variables u1-u9 represent base usage for a gene.
2.3.. COA and PR2-Plot
COA is a classical technique to reduce the dimensionality of the dataset by transforming to a new set of variables (the principal components) to summarize the feature of the data [15]. The new set of variables is derived from the linear combination of the original variables. The first principal axis is chosen to maximize the standard deviation of the derived variable and the second principal axis is the direction to maximize the standard deviation among directions uncorrelated with the first, and so forth. For details about this method, refer to Dillon and Goldstein [17]. The results of a COA are viewed graphically, usually by plotting the coordinates of all genes along the first eigenvectors [15]. Here, COA is adopted to show the different base and codon usage of genes located on the leading and lagging strands. COA is computed on the variables u1-u9, 59 codon counts and 59 RSCU values, respectively.
According to the parity rule 2, the average nucleotide composition is theoretically expected to be A = T and G = C within each strand when there are no strand-specific biases in the substitution rates between the two strands of DNA. Deviations from these equalities are therefore evidences for an asymmetry in mutation and/or selection between the two strands. PR2-plot, proposed by Lobry and Sueoka [4], donates G/(G+C) against A/(A+T) at the 3rd codon position in genes and it could reflect the deviations. Here, we adopt PR2-plot method to show nucleotide composition bias of gene in E. canis genome.
3.. RESULTS AND DISCUSSION
3.1.. Separate Base/Codon Usage Revealed by Three Types of COA
For each of the 925 genes in E. canis genome, we calculate 59 codon counts (excluding three stop codons and codons encoding for Met and Trp), 59 RSCU values and u1-u9. Each of these three types of variables for a gene corresponds to a point in 59-D, 59-D and 9-D space, respectively. In order to visualize the distribution of 925 mapping points in high dimensional space, project them to a 2-D plane spanned by the first and second principal axes by using the COA method. Figs. (1a), (b) and (c) show the position of the genes on the 2-D principal plane after COA on codon counts, RSCU values and u1-u9, respectively. As can be seen from each of the figure, all the genes are divided into two quite distinct clusters with little overlap, which indicates that genes in the two clusters have different codon and base usages.
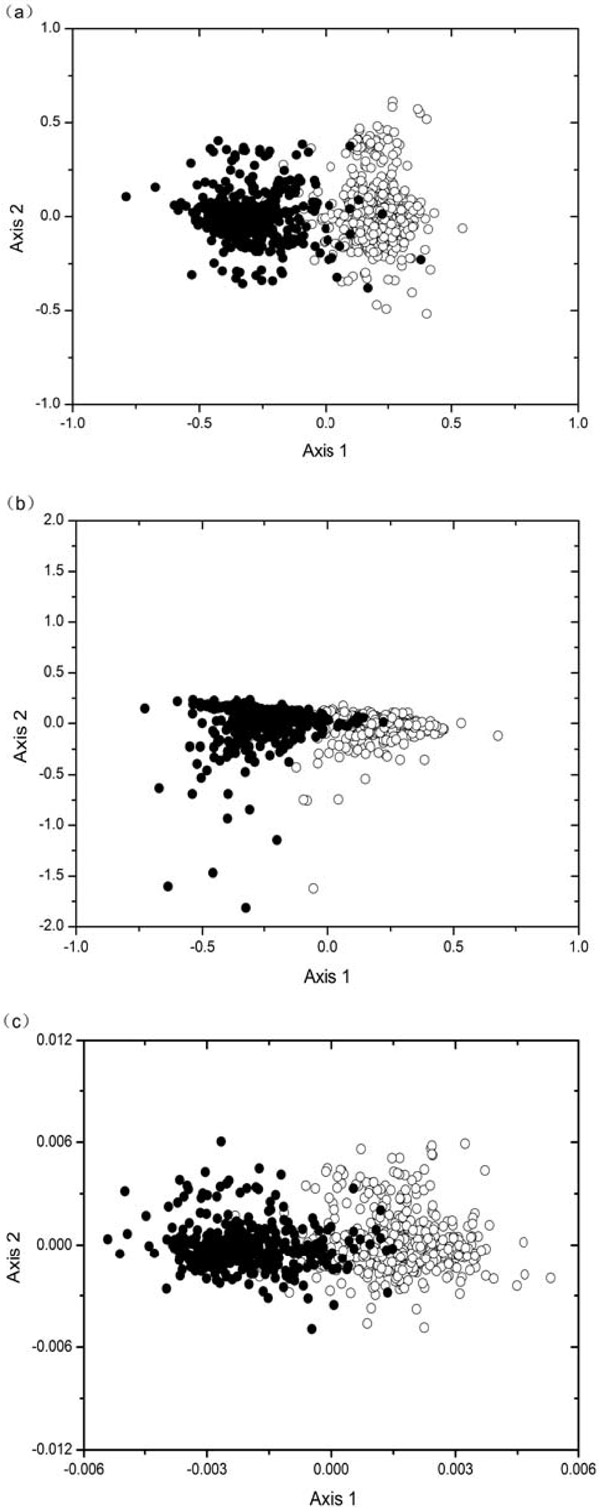
The distribution of points based on the two most important axes by using the COA for 925 genes of the E. canis genome. Genes located on leading strands are denoted by open circles, whereas filled circles indicate lagging strand genes. (a) COA on codon counts; (b) COA on RSCU values; (c) COA on nine variables (u1-u9) of phase-specific Z curve.
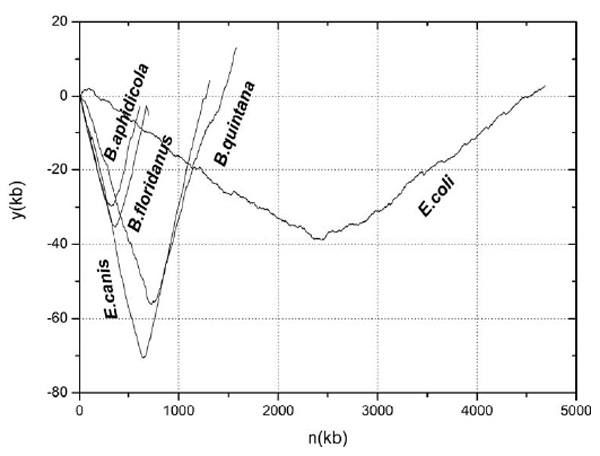
Y component curves for four representatives of 11 obligate intracellular bacteria and that for the free-living bacterium E.coli K12. To allow convenient observation and direct comparison, the first base of the chromosome is shifted to the origin of replication for each genome.
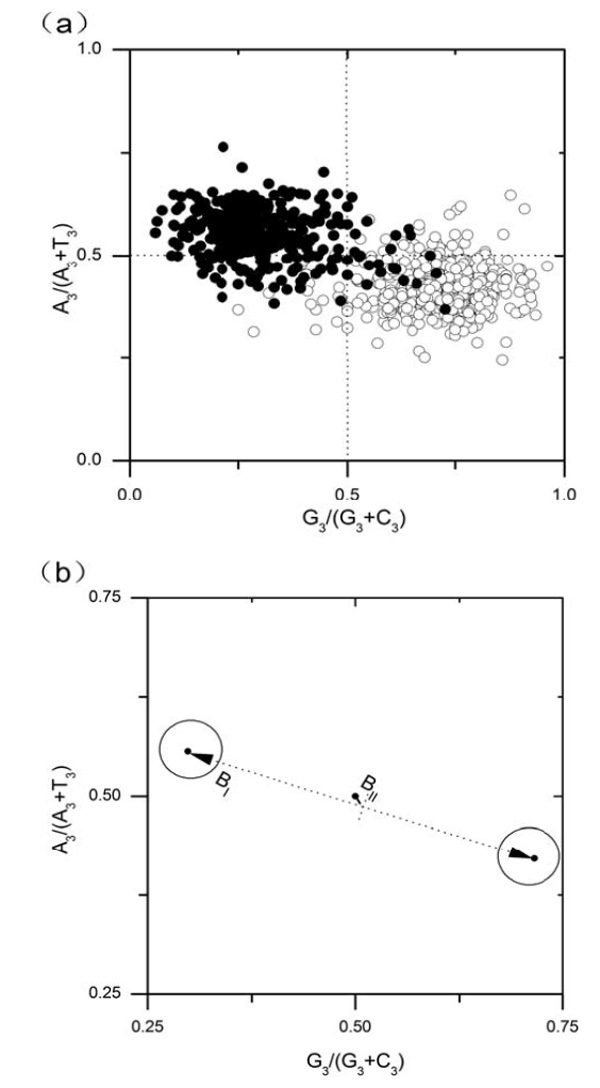
PR2-plot for E.canis chromosome. Genes located on the leading strands are denoted as open circles, whereas filled circles indicate lagging strand genes. In figure (a), each gene is denoted by a circle and whereas in (b), each category of genes, as a whole, is denoted by a larger circle.
On inspection, it is found that the two groups correspond to the genes that are transcribed either in the leading strands or in the lagging strands, respectively. This phenomenon, i.e., separate base/codon usages of the genes in the two replicating strands, is similar to that observed previously in genomes of B. burgdorferi, Treponema pallidum, Chlamydia trachomatis, B. aphidicola, B. floridanus, Bartonella henselae, Bartonella quintana, Tropheryma whipplei, Chlamydia muridarum and Lawsonia intracellularis [10-13]. All of the ten bacteria and here the E. canis are all obligate intracellular. They have extreme strand composition bias and thus their genes have two separate base/codon usages.
3.2.. Two Common Genomic Characters Revealed by y Component of Z Curve
Eleven obligate intracellular bacteria have been found to have separate base/codon usages according to whether genes located on the leading or lagging strands. Investigating the common genomic characters of them may be interesting and important. In previous works [12], we have tried to do this by using Z curve method shown in Equation (1). In this work, only y component curve is needed.
In Fig. (2), y component curves are shown for four representatives of 11 obligate intracellular bacteria. For convenient observation, the other 7 bacteria are not shown but they have similar y component curves. For comparison, y component curve of E. coli K12 chromosome is also shown. In E. coli, there also exists strand specific composition bias, whereas it is not strong enough to generate separate codon usages. From this figure, we could make the following two observations. Firstly, changing rates of y component curves for the four representative obligate intracellular bacteria are much higher than that of the free-living E. coli. According to equation (1), the y component curve represents the cumulative excess of G over C plus T over A. Therefore, comparison between y component curves of the two groups of bacteria suggests that 11 obligate intracellular bacteria have stronger strand composition bias. Secondly, all of the y component curves for four bacteria are much smoother than that of E. coli. In y component curve for the latter, there are many prickles along the chromosome, whereas not so many for the former. As shown in Grigoriev [18], prickles in the chromosome diagrams usually denote sequence inversions or direct translocations to another half of a chromosome, or integration of foreign DNA into the chromosome. In other words, chromosome rearrangements usually are exhibited as little prickles in the y component curves. Therefore, we could make the conclusion that the 11 obligate intracellular chromosomes are highly stable and have very few rearrangements. As suggested by Rocha [7] and others, stronger strand composition bias and lower rearrangements frequency are just the most likely reasons for the appearance of separate base/codon usages in some obligate intracellular bacteria. Our results confirmed their speculations.
3.3.. Using PR-2 Plots to Differentiate Replication and Transcription Associated Biases
As mentioned above, composition bias among genes is very strong in E. canis genome. These biases are induced by asymmetric replication mutation pressure or by asymmetric transcription/translation-associated mutation/selection pressure. To differentiate replication-induced bias and transcription/translation associated bias, PR2-plots for single gene and gene clusters are drawn in Figs. (3a) and (b), respectively. As can be seen from figure (a), the strand bias of G/C, which could be reflected by the values of the horizontal axis of the plot [4], is much stronger than that of T/A (reflected by the vertical axis). In figure (b), BI denotes the distance between the centers of two clusters of genes, whereas BII denotes the distance from the origin of coordinates system to the midpoint of two centers. In fact, BI means the extent of replication-induced bias and BII reflect that of transcription/translation-associated bias [4]. Values of BI , BII are 0.438 and 0.013, respectively. Therefore, replication effect on composition bias of genes is much higher than that of transcription/translation effect in E. canis genome.
3.4.. Why do We Combine Multiple Methods in This Work?
In this work, we have employed three methods to reveal the strong strand composition bias in E. canis. Merits and advantages of each method are described as follows.
Plane plots after COA could intuitively visualize the difference of nucleotide composition between genes located on the two replicating strands. As long as strand composition bias is strong enough to affect base/codon usage, it will be exposed to COA plots. Three types of COA are employed in this work. Contents reflected by them are different. COA on u1-u9 reflects distribution pattern of base usages of genes. COA on codon reflects distribution of absolute codon numbers, whereas COA on RSCU reflects distribution of relative synonymous codon usage [15]. By using three types of COA plots in this work, we could make the conclusion that base usage, absolute codon usage, relative codon usage are all separated between genes on the two replicating strands in E. canis genome. Furthermore, division of two clusters of genes in Figure (a) is significantly clearer than that in Figure (b). In fact, COA on codon counts also has the advantage over RSCU when analyzing separate codon usages in L. intracellularis genome [13]. On the other hand, division of two clusters in Figure (c) is slightly clearer than that in Figure (a). This is consistent with previous observation that base usages are more sensitive than codon usages for discriminating genes on the two replicating strands in four bacterial genomes.
We used y component curves of the Z curve method to investigate the common genomic characters among 11 obligate intracellular bacteria. After comparison with E. coli K12, they are found to have two common genomic characters, i.e. stronger strand composition bias and lower rearrangement frequency. Therefore, Z curve is a useful tool to analyze the global genomic characters of bacteria. By using it, important results may be obtained.
PR2-plot [4], as a diagram tool, could not only differentiate replication-induced bias and transcription/translation-associated bias but also evaluate the relative strength between bias of G/C and that of T/A.
In brief, each method has its merits. Complete, reliable and interesting results (or conclusions) may be obtained when combining the multiple methods to analyze strand composition bias in bacteria. The methods could be applied to other sequenced microbes, particularly obligate intracellular bacteria.
ACKNOWLEDGEMENTS
We are grateful to the anonymous reviewers for their valuable suggestions and comments, which have led to the improvement of this paper. The present study was supported by Doctoral Fund of Ministry of Education of China (grant 20070614011) and by Committee of Science and Technology of Sichuan Province (grant 2008JY0053).

