All published articles of this journal are available on ScienceDirect.

Purification of Protease from Pseudomonas thermaerum GW1 Isolated from Poultry Waste Site
Abstract
An extracellular protease was purified from Pseudomonas thermaerum GW1 a new strain identified by morphological, biochemical and 16S rDNA sequencing. It was isolated from soil of Poultry waste site at Ghazipur near Ghaziabad, Delhi. The strain produces extra cellular protease in the culture media that was maintained at 37°C, 140 rpm. The media was harvested for protease after 48 hrs of incubation at 37°C in basal media supplemented with 1% casein. We report 6.08 fold purification of enzyme following ammonium sulphate precipitation and DEAE-cellulose chromatography. The molecular weight of the enzyme was estimated to be approximately 43,000 daltons as shown by casein zymography studies. The optimum pH for the proteolytic activity was pH 8.0 and enzyme remained stable between pH 5 -11 at 60°C. Interestingly Mn2+ (5mM) activated enzyme activity by 5 fold, while Cu2+, Mg2+and Ca2+ moderately activated enzyme activity, where as Zn2+, Fe2+ and Hg2+ inhibited enzyme activity. The protease produced was stable in presence of 50 % (v/v) ethylacetate and acetone. Isopropanol, methanol and benzene increased protease activity by 2.7, 1.3 and 1.1 fold respectively but was inhibited in presence of glycerol and DMSO. This organic solvent-stable protease could be used as a biocatalyst for enzymatic peptide synthesis
INTRODUCTION
Proteases covers up to 60% of total enzyme market and are valuable commercial enzyme that have biotechnological as well as industrial applications however the present known proteases are not sufficient to meet most of the industrial demands It is desirable to have new proteases with novel properties from different sources. Alkaline proteases hold a great potential for application in the detergent and leather industries [1-3] and are also reported to have been isolated from microbes, plants and animals.
Proteases from plant sources have application in food industry [4]. Previous studies in our lab have shown that proteases from senesced leaves of Lantana camara can have application in detergent industry as the enzyme is thermostable [5]. Microbes are the preferred source of proteases because of their rapid growth, and the ease with which they can be genetically manipulated to generate new enzymes with altered properties [6, 7]. Proteases have been purified and characterized from several bacteria [8-12]. However there are only few reports on Pseudomonas thermaerum, Yang et al., [13] have identified two strains of Psudomonas thermaerum isolated from activated sludge that could use lignin as sole carbon source and excrete peroxidases. A novel antimicrobial peptide (30 kDa) produced by a this bacterium isolated from the effluent pond of a bovine abattoir showed inhibition to a broad range of indicator strains, including pathogenic and food spoilage bacteria such as Listeria monocytogenes, Bacillus cereus, Staphylococcus aureus has been reported by Fontoura et al., [14]. The partially purified antimicrobial substance remained active over a wide temperature range and was resistant to all proteases. S28 strain isolated from Nanyang oil field was identified to be as P. thermaerum based on homology studies and could degrade 82.02% of phenenthrene within 10 days [15].
In our attempt to screen for microbial isolate that can provide stable enzyme, soil sample of the poultry waste site which is rich in organic waste was selected. In this investigation, we report the production and effect of pH, temperature, metal ions and substrate concentration on protease secretion whereby the culture condition were manipulated for maximum protease production. Pseudomonas thermareum GW1 was isolated from soil of poultry waste site and it extracellularly secreted this protease which was purified to homogeneity, characterized and found to be stable in the presence of several organic solvents. Ours is the first report that shows extracellular production of proteases from Pseudomonas thermaerum GW1 strain.
MATERIALS AND METHODS
Isolation and Screening of Protease-Producing Strains
Soil was collected in sterilized sampling bags of Ghazipur poultry waste site, India. The microbe responsible for the production of protease was identified. The classification was based on gram strain (-), catalase (+) and oxidase (+) reactions, using morphological and biochemical characteristics based on Bergey’s Manual of Determinative Bacteriology [16]. Soil was selected from areas where naturally degraded feather were seen. The samples were brought to the laboratory and processed for the analysis the same day. Soil samples were suspended in basal media and kept for growth at 37°C for 6 days. At regular intervals of 6 hrs the activity of protease was measured and sample showing maximum activity was screened for protease producing strains. Samples of repeated batch cultures were plated on skim milk agar. After 24–48 hrs at 37°C, colonies which exhibited the largest cleared zones were selected and was further incubated in cultivation media for further 48 hrs and checked for protease production.
Identification of Protease Producing Bacteria
The isolate GW1 was identified originally as a strain of Pseudomonas by our laboratory based on Morphological, Physical, Biochemical characteristics and the single colony was subcultured on bacterial culture plates supplemented with casein. The culture was sent for identification till species level to Bangalore GeneI India by partial 16S r DNA sequence analysis.
Production of Enzyme in Cultivation Media
The Basal media for protease production composed of (g L-1): Peptone, 5; Glucose, 10; NaCl, 0.5; CaCl2.2H2O, 0.1; K2HPO4, 0.3; KH2PO4, 0.4; MgSO4 · 7H2O, 0.1; and yeast extract, 5. The pH was maintained at 7.5. Microbes were allowed to grow in 500 ml conical flask containing 50 ml of the culture media that was maintained at 37°C at 140 rpm. 5% (v/v) of the 20 hrs old culture was inoculated in cultivation media [17].
- Tryptic soy broth (TSB)
- Gelatin (1%) + basal medium for protease production
- Casein (1%)+ basal medium for protease production
- Skim milk powder (1%)+ basal medium for protease production
- Pigeon feathers (1%)+ basal medium for protease production
The above cultivation media were checked for enzyme activity at regular intervals of 6 hrs by the modified method of Tsuchida et al. [18].
Protease Purification
The bacterial strain was grown for 48 hrs at 37°C in the selected cultivation media. The culture medium was centrifuged at 10,000 rpm for 10 min at 4°C and the cell-free supernatant was precipitated with 0-60% ammonium sulfate. The precipitate was collected by centrifugation and dissolved in a small volume (1/50) of 10 mM Tris-HCl buffer (pH 8.0), and dialyzed against 4 liters of same buffer for 12 hrs at 4°C. This step was repeated twice. The dialyzed enzyme preparation was applied on a DEAE-cellulose column (2 X 24 cm) pre-equilibrated with 10 mM Tris-HCl (pH 8.0). The unadsorbed protein fraction was eluted with the same buffer (150 ml). The enzyme was eluted with a gradient of 2mM and 4 mM NaCl in the same buffer at a flow rate of 1ml/min. Active fractions that contained (80%) of the enzyme activity were pooled, and subsequently used for characterization. All steps were conducted at 4°C.
Determination of Protease Activity
Protease activity was assayed by a modified method of Tsuchida et al. [18] by using casein as substrate. 100 μl of enzyme solution was added to 900 μl of substrate solution (2 mg/ml (w/v) casein in 10 mM Tris–HCl buffer, pH 8.0).The mixture was incubated at 45°C for 30 min. Reaction was terminated by the addition of an equal volume of 10% (w/v) chilled trichloroacetic acid then the reaction mixture was allowed to stand in ice for 15 min to precipitate the insoluble proteins. The supernatant was separated by centrifugation at 10,000 rpm for 10 min at 4°C; the acid soluble product in the supernatant was neutralized with 5 ml of 0.5 M Na2CO3 solution. The colour developed after adding 0.5 ml of 3-fold-diluted Folin–Ciocalteau reagent was measured at 660 nm. All assays were done in triplicate. One protease unit is defined as the amount of enzyme that releases 1 µg of tyrosine per ml per minute under the above assay conditions. The specific activity is expressed in the units of enzyme activity per milligram of protein.
Protein Concentration
Protein concentration was determined by the method of Bradford [19] with bovine serum albumin as standard.
Polyacrylamide Gel Electrophoresis and Zymogram
SDS-PAGE was performed on a slab gel containing 10% (w/v) polyacrylamide by the method of Laemmli [20]. Casein zymography was performed in polyacrylamide slab gels containing SDS and casein (0.12% w/v) as co-polymerized substrate, as described by Choi et al., [21]. After electrophoresis, the gel was incubated for 30 minutes at room temperature on a gel rocker in 50 mM Tris-Cl (pH 7.4), which contained 2.5% Triton X-100 to remove SDS. The gel was then incubated in a zymogram reaction buffer (30 mM Tris-HCl, pH 7.4, 200 mM NaCl and 10 mM CaCl2) left at 37°C for 12 hrs on rocker shaker.The gel was stained with Coomassie brilliant blue (0.5% w/v) for 30 min. The activity band was observed as a clear colourless area depleted of casein in the gel against the blue background when destained in 10% methanol and 5% acetic acid for a limited period of time.
Effect of pH on Enzyme Activity
Effect of pH on the purified enzyme activity was measured at various pH ranges (3.0 – 12). Reaction mixtures were incubated at 45°C for 30 min and the activity of the enzyme was measured as described previously.
Effect Of Temperature On Enzyme Activity And Stability
The activity of the enzyme was determined by incubating the reaction mixture at different temperatures ranging from 20, 30, 40, 50, 60, 70 and 80°C were studied. The activity of the enzyme was measured as described previously.
Effect of Various Metal Ions on Protease Activity
The effects of metal ions on enzyme activity (e.g., Ca2+, Mg2+, Fe2+, Mn2+, Zn2+, Hg2+, and Cu2+ [5 mM]) were investigated by adding them to the reaction mixture and pre-incubated for 30 min at 45°C pH 10.0. The activity of the enzyme was measured as described previously.
Effect of Organic Solvents on the Protease Stability
The organic solvents used were Methanol, Ethyl acetate, Benzene, Glycerol, Sucrose, Toluene, Acetone, Hexane, DMSO, Isopropanol and Ethanol. In the stability test, 1.0 ml of organic solvent (100% v/v) was added to 1 ml of the reaction mixture and pre incubated at 37°C for 30 min. The remaining proteolytic activity was measured as described previously. Stability was expressed as the remaining proteolytic activity relative to the solvent-free controls (0%, v/v).
RESULTS AND DISCUSSION
Isolation and Identification of Protease-Producing Bacterial Strains
Soil samples were analyzed for isolation of proteolytic bacterial cultures. Screening of microorganisms that produced protease was done on cultures isolated from soil of Ghazipur poultry waste site. Organic waste such as feathers and other poultry waste is essentially composed of proteins. Protease producing strains were selected by growth on skim milk agar, as described in Methods. Among the cultures tested, the laboratory isolate GW1 showed highest zone of clearance. The purity of the isolated bacteria was ascertained through repeated streaking (Fig not shown).
Microscopic observation of the isolate showed a non sporulating gram negative rods, the bacterium grew aerobically and formed typical blue green, flat, large, grape like odour colonies. The strain showed positive reaction for catalase, oxidase, citrate, nitrate, motility, and production of pyoveridin and pyocyanin. Negative reactions were observed for indole, urea and starch hydrolysis (Table 1). These phenotypic characteristics based on Bergey’s Manual of Determinative Bacteriology [16] suggest the Pseudomonadaceae family genus Pseudomonas.
Morphological and Biochemical Characteristics of Isolate
| Morphological and Biochemical Characteristics | Results |
|---|---|
| Colony morphology | Irregular,Undulated,slimy and flat |
| Texture | Shiny, smooth |
| Odour | Sweet grapey |
| Gram staining | Gram Negative rods |
| Spores | - |
| Aerobic growth | + |
| Motility | + |
| Catalase | + |
| Oxidase | + |
| Glucose | - |
| Lactose | - |
| Sucrose | - |
| Methyl Red | - |
| V-P test | Indole |
| Citrate | + |
| Nitrate reduction | + |
| Urea Test | - |
| Starch Hydrolysis | - |
| Pseudomonas agar P | + blue green pigment |
| Cetrimide Agar | +, Pyocyanin (blue green pigment) production |
| Identification of organism | Genus Pseudomonas |
Table gives the Alignment view of the Sequence of the Isolated Microbe Using Combination of NCBI GenBank and RDPDatabase Using 10 Examples. Nearest Homolog was Found to be Pseudomonas Aeruginosa Strain EK1 (Accession No.FJ685995)
| Alignment View | ID | Alignment Results | Sequence Description |
|---|---|---|---|
 |
PGW1B | 0.81 | Studied sample |
 |
FJ685995 | 0.83 | Pseudomonas thermaerum strain EKi |
 |
FJ816019 | 0.83 | Pseudomonas aeruginosa strain EK1 |
 |
FJ948174 | 1.00 | Pseudomonas aeruginosa strain WJ-1 |
 |
FJ864676 | 0.99 | Pseudomonas aeruginosa strain pp1a |
 |
EU352760 | 1.00 | Pseudomonas aeruginosa strain NK 2.1B-1 |
 |
EU331416 | 1.00 | Pseudomonas aeruginosa strain pY11T-3-1 |
 |
EU331416 | 1.00 | Pseudomonas aeruginosa strain pY11T-3-1 |
 |
EU099381 | 0.99 | Pseudomonas sp. J16 |
 |
AB305018 | 0.99 | Pseudomonas aeruginosa strain PA1 |
 |
EU603683 | 0.99 | Pseudomonas aeruginosa strain XJTUMS3 |
 |
EF551040 | 0.93 | Pseudomonas sp. GZ1 |
Indicates Nucleotide Similarity (Above Diagonal) and Distance (Below Diagonal) Identities Between the Studied Sample ‘PGW1B’ and Ten other Closest Homologs Microbe
| Distance Matrix | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | ||
| FJ948174 | 1 | --- | 0.996 | 0.949 | 0.999 | 0.999 | 1 | 0.949 | 0.999 | 0.960 | 0.996 | 0.940 |
| EU331416 | 2 | 0.004 | --- | 0.944 | 0.995 | 0.995 | 0.996 | 0.944 | 0.996 | 0.955 | 1 | 0.936 |
| FJ685995 | 3 | 0.051 | 0.056 | 0.051 | 0.002 | --- | 0.999 | 0.949 | 0.948 | 0.906 | 0.944 | 0.982 |
| EU099381 | 4 | 0.001 | 0.005 | 0.052 | --- | 0.999 | 0.999 | 0.948 | 0.999 | 0.959 | 0.995 | 0.939 |
| FJ864676 | 5 | 0.001 | 0.005 | 0.051 | 0.002 | --- | 0.999 | 0.949 | 0.999 | 0.959 | 0.995 | 0.940 |
| AB305018 | 6 | 0.000 | 0.004 | 0.051 | 0.001 | 0.001 | --- | 0.949 | 0.999 | 0.959 | 0.995 | 0.940 |
| FJ816019 | 7 | 0.051 | 0.056 | 0.000 | 0.052 | 0.051 | 0.051 | --- | 0.948 | 0.906 | 0.944 | 0.982 |
| EU603683 | 8 | 0.001 | 0.004 | 0.052 | 0.002 | 0.002 | 0.001 | 0..052 | --- | 0.959 | 0.996 | 0.940 |
| EF551040 | 9 | 0.041 | 0.045 | 0.094 | 0.041 | 0.041 | 0.994 | 0.094 | 0.041 | --- | 0.955 | 0.894 |
| EU352760 | 10 | 0.004 | 0.000 | 0.056 | 0.005 | 0.005 | 0.004 | 0.056 | 0.004 | 0.0.45 | --- | 0.936 |
| PGW1B | 11 | 0.060 | 0.064 | 0.018 | 0.061 | 0.060 | 0.060 | 0.018 | 0.060 | 0.106 | 0.064 | --- |
Summary of Purification Steps of Alkaline Protease from Pseudomonas thermaerum
| Purification Step | Total Protein(mg) | Total Enzyme Activity(U) | Specific Activity(U/mg) | % Recovery | Purification Fold |
|---|---|---|---|---|---|
| Culture supernatant | 117.4 | 2645 | 22.6 | 100 | 1 |
| (NHᶺ)2SOᶺ Precipitation, dialyzed (0-60%) | 36.70 | 1940.40 | 52.87 | 73.3 | 2.33 |
| DEAE cellulose | 1.79 | 246.15 | 137.54 | 9.3 | 6.08 |
Summary of Purification Steps of Alkaline Protease from Pseudomonas thermaerum
| Metal Ion (5 mM) | Relative protease Activity (%) |
|---|---|
| Control CuSO4 MnSO4 HgC2 FeCl3 CaCl2 MgCl2 ZnSO4 | 100 109.57 546.80 69.14 1.0 126.59 174.46 80.85 |
Stability of Enzyme in the Presence of Glycerol
| Glycerol (%) | Relative Protease Activity (%) |
|---|---|
| Control (without glycerol) | 100 |
| 10 | 62.6 |
| 20 | 55.2 |
| 40 | 25.2 |
| 60 | 23.9 |
| 80 | 17.3 |
| 100 | 0 |
Strain Identification by 16S rDNA Sequencing
The GW1 strain was identified to be as Pseudomonas thermaerum as predicted by 16S rDNA studies. Studying the Alignment view of the sequence of the isolated microbe using combination of NCBI GenBank and RDP database using 10 examples and nucleotide similarity. Nearest homolog was found to be Pseudomonas aeruginosa strain EKi (Accession No. FJ685995). The details are given in Table 2, 3 respectively.
The sequence of the isolate GW1 was submitted to the GenBank (Accession Number GU951516)” and based on nucleotide homology and phylogenetic analysis it is found to have close similarity to Pseudomonas thermaerum strain EKi (GenBank Accession Number: F3816019) (Fig. 1).

Phylogenetic tree made in MEGA 3.1 software using neighbor joining method.
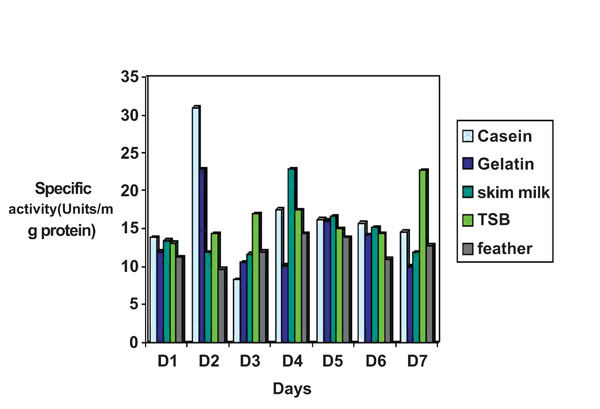
Study of effect of various nutrient sources on protease production by Pseudomonas thermaerum GW1. The culture was grown for production of protease as described in “Methods”. 1% of the above mentioned nutrient sources were added to basal media. pH of the media was adjusted to 7.5.
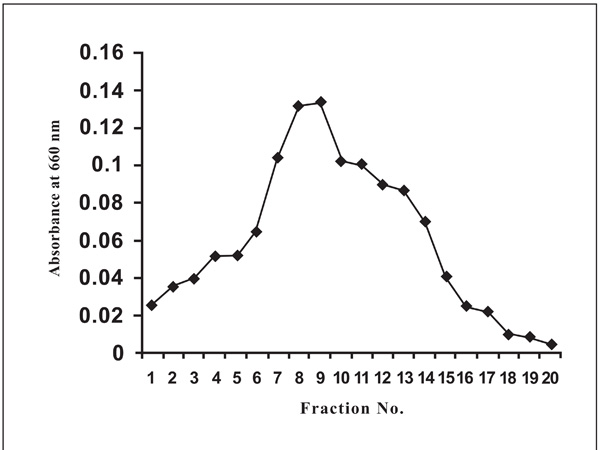
The bound protease on DEAE column was eluted with 0.2 and 0.4 M NaCl (in 10 mM Tris–HCl buffer, pH 8.0) Fraction of 0.4M NaCl showed a single peak of caseinase activity.
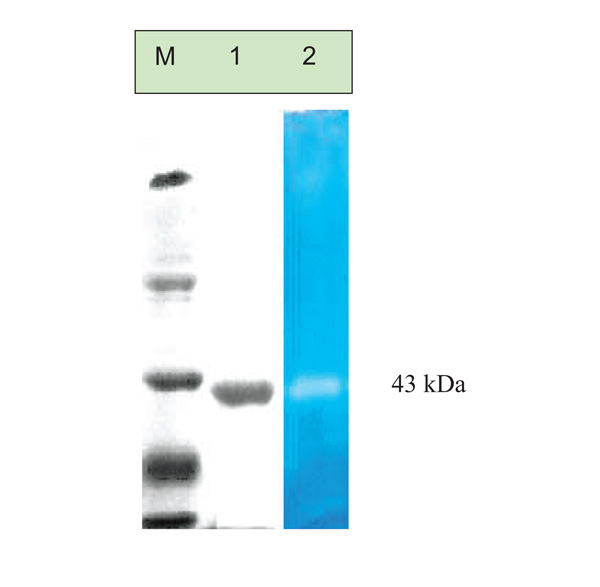
Acitvity gel electrophoresis of the purified protease, M: molecular weight markers, Lane 1: purified protease on 10% SDS-PAGE and Lane 2: Zymography of purified protease from Pseudomonas thermaerumGW1. Zymography was done by the method of Choi et al.

Effect of pH on the activity of the purified Protease from Pseudomonas thermaerum GW1 pH optima was measured by incubating the enzyme with the substrate at different pH values at 45°C. The maximum activity obtained at pH 8.0 was considered as 100% activity. The treated enzyme solution was cooled rapidly in ice and the relative activity was measured under standard condition.
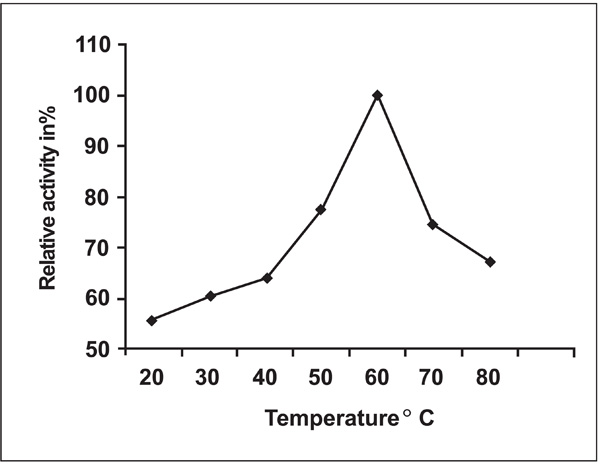
Effect of temperature on protease activity from Pseudomonas thermaerum GW1. The relative activity was defined as the percentage of activity detected with respect to the maximum protease activity. The maximum activity obtained at temperature 60°C was considered as 100% activity. The treated enzyme solution was cooled rapidly in ice and the relative activity was measured under standard condition.
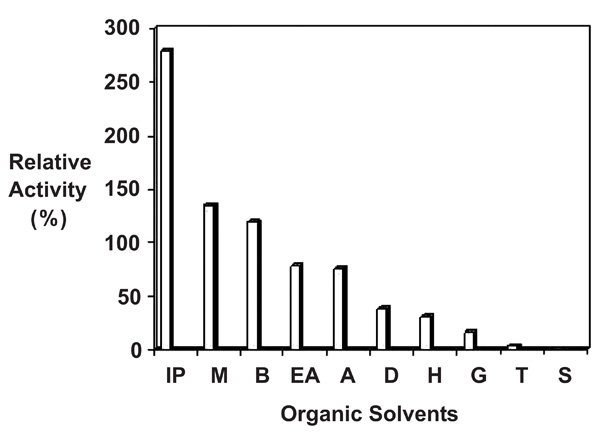
Figure denotes Influence of different solvents on activity of the purified protease from Pseudomonas thermaerum GW1: purified enzyme (10µg) was preincubated with 100% v/v of various solvents vis Isopropanol (IP), Methanol (M), Benzene (B), Ethylacetate (EA), Acetone (A), DMSO (D), Hexane (H), Glycerol (G), Toluene (T), Sucrose (S) at 37°C for 30 minutes. The control (without organic solvent) and treated enzyme solution was cooled rapidly in ice and the relative activity was measured under standard condition as described in methods. Note: the relative enzyme activity of control fraction without organic solvent (0% v/v) was denoted as 100% The relative activity of treated fractions was defined as the percentage of activity detected with respect to the control fraction.
Protease Production and Effects of Different Parameters
Protease production was tested at various time interval (1–7 days) and influence of addition of various nutrient sources (TSB, Gelatin, Casein, Skim milk, Pigeon feathers) were evaluated in relation to enzyme yield. Pseudomonas thermaerum strain GW1 grew in five nutrient sources and produced protease. The highest protease production 32 units/mg occurred in Basal medium supplemented with casein whereas lowest in basal medium supplemented with Pigeon feathers 9.7units/mg protein after 48 hrs of cultivation (Fig. 2).
Purification of Protease
The extracellular protease produced by Pseudomonas thermaerum strain GW1 was purified in two steps by 0-60% ammonium sulphate precipitation followed by anion exchange chromatography on DEAE- cellulose resin (Fig. 3). The recovered active fraction from 0-60% ammonium sulphate of culture broth was adsorbed on the DEAE- cellulose matrix. The bound protease was eluted with 0.2 and 0.4 M NaCl (in 10 mM Tris–HCl buffer, pH 8.0). The protease was purified 6.08 fold and about 9.3% of the total activity units was recovered. The specific activity of the purified enzyme was 137.54 units/mg. The purified enzyme could be stored in 10 mM Tris-HCl buffer, pH 8.0, at -80ºC for 3 months without any apparent loss of activity. The results of purification of protease from Pseudomonas thermaerum strain GW1 are summarized in Table 4.
SDS-PAGE and Zymogram Analysis
The DEAE fraction was analysed on SDS PAGE (10%), showed presence of single band indicating a homogeneous preparation. The enzyme has a low molecular weight of approximately 43Kda (Fig. 4 Lane 2). Zymogram activity staining also revealed one clear zone of proteolytic activity against the blue background for purified sample at corresponding positions in SDS-PAGE (Fig. 4 Lane 3).
pH Optimum and pH Stability
Activity of the enzyme was determined at different pH ranging from 3.0-12.0. The optimum pH recorded was 8.0 for protease activity. Protease activity was found to be stable in the alkaline range starting from the pH 5-11 at 45°C (Fig. 5).
Temperature Optimum and Thermal Stability
The thermal stability of the enzyme was also tested at different temperatures 20°C, 30°C, 40°C, 50°C, 60°C, 70°C and 80°C on incubation for 60 minutes (Fig. 6). The optimum temperature recorded was at 60°C for protease activity and Protease activity was found to be stable in the temperature range from 40°C - 70°C. The enzyme activity gradually declined at temperatures beyond 70°.
Effect of Metal Ions
Fe2+ has a strong inhibitory effect, whereas Zn2+ and Hg2+ have mild effects on protease activity. Interestingly Mn2+ strongly activated enzyme activity by 5 fold (Table 5).
Effect of Organic Solvents on the Protease Stability
Ten organic solvents were used to study the effect on protease activity. As shown in (Fig. 7) the protease has ability to act in the presence of solvents in reaction system. The enzyme retained 78% and 75 % of activity in the presence of ethylacetate and acetone respectively. The presence of isopropanol, methanol and benzene increased the activity of isolates GW1 by 2.7, 1.3 and 1.1 fold, respectively. Enzyme lost 60% of total activity in presence of DMSO and hexane and was not stable in the presence of glycerol, toluene and sucrose. Ogino et al., [22] have reported importance of disulfide bonds for stability of the protein in presence of solvents. Jorden et al., [23] have reported that 61% of the activity of HIV1 protease was lost in presence of 12% Me2SO; similar results were also reported from protease thrombin [24]. This loss of hydrolytic activity over time as reported by the authors is not due to slow dissociation of enzyme dimer into inactive monomer. The activity of HIV1 is also sensitive to glycerol and the hydrolytic efficiency of this enzyme decreases with the increasing concentration of glycerol.
Table 6 reveals that if the concentration of glycerol is as low as10%v/v. then there is 38% decrease in Protease activity whereas 45 %decrease in enzyme activity is reported in presence of 20%v/v glycerol.
Ours is the first report that shows extracellular production of proteases from Pseudomonas thermaerum. Protease from GW1 strain lost its activity in the presence of glycerol, sucrose and metal ion iron.
CONCLUSIONS
Enzyme activity from Pseudomonas thermaerum is lost in the presence of glycerol and sucrose. The buffer best suited for Pseudomonas thermaerum protease should minimize the use of glycerol and sucrose during dialysis.
So far, several well-known proteases such as thermolysin, papain, and chymotrypsin have been used as biocatalysts of peptide synthesis in the presence of organic solvents. Protease from Pseudomonas thermaerum retained its activity in organic solvents; these results are in line with many studies that report protease stability in the presence of organic solvents [25-29] promising potential industrial application of protease from Pseudomonas thermaerum.
ACKNOWLEDGMENT
We are thankful to Department of Biotechnology, Jaypee Institute of Information Technology (Deemed University) Noida, India for providing infrastructure facilities for this study.

