All published articles of this journal are available on ScienceDirect.

Infective Endocarditis: Identification of Catalase-Negative, Gram-Positive Cocci from Blood Cultures by Partial 16S rRNA Gene Analysis and by Vitek 2 Examination
Authors Info & Affiliations
Abstract
Streptococci, enterococci and Streptococcus-like bacteria are frequent etiologic agents of infective endocarditis and correct species identification can be a laboratory challenge. Viridans streptococci (VS) not seldomly cause contamination of blood cultures. Vitek 2 and partial sequencing of the 16S rRNA gene were applied in order to compare the results of both methods.
Strains originated from two groups of patients: 149 strains from patients with infective endocarditis and 181 strains assessed as blood culture contaminants. Of the 330 strains, based on partial 16S rRNA gene sequencing results, 251 (76%) were VS strains, 10 (3%) were pyogenic streptococcal strains, 54 (16%) were E. faecalis strains and 15 (5%) strains belonged to a group of miscellaneous catalase-negative, Gram-positive cocci. Among VS strains, respectively, 220 (87,6%) and 31 (12,3%) obtained agreeing and non-agreeing identifications with the two methods with respect to allocation to the same VS group. Non-agreeing species identification mostly occurred among strains in the contaminant group, while for endocarditis strains notably fewer disagreeing results were observed.
Only 67 of 150 strains in the mitis group strains obtained identical species identifications by the two methods. Most VS strains belonging to the groups of salivarius, anginosus, and mutans obtained agreeing species identifications with the two methods, while this only was the case for 13 of the 21 bovis strains. Pyogenic strains (n=10), Enterococcus faecalis strains (n=54) and a miscellaneous group of catalase-negative, Gram-positive cocci (n=15) seemed well identified by both methods, except that disagreements in identifications in the miscellaneous group of strains occurred for 6 of 15 strains.
INTRODUCTION
Viridans streptococci (VS), enterococci and Streptococcus-like bacteria are frequent etiologic agents of infective endocarditis and correct species identification can be a big laboratory challenge [1]. Diagnostic tools for identification of bacteria have developed dramatically in the last decades. These techniques, especially sequencing of the genes coding for rRNA and other genes, have led to revolutionary insights into the phylogeny and taxonomy of streptococci and related taxons [2]. Accurate identification of ß-hemolytic or pyogenic streptococci is to a large extent done by phenotypic reactions, whereas newer molecular techniques including sequencing of genes are more and more often used for their typing [2]. Accurate identification of strains belonging to viridans streptococci is prerequisite for understanding the pathogenesis of these opportunistic infections, especially with regards to infective endocarditis [3].
The genus Streptococcus consists of more than 65 validly published species, and the taxonomic classification of these members is not well defined, but they are divided into: the pyogenic group and the VS, which are grouped into 1) the anginosus group, formerly called “ S. milleri”, 2) the mitis group, 3) the salivarius group, 4) the mutans group, 5) the bovis group and 6) other undifferentiated streptococci [4]. Knowledge on identification and relationship among VS strains increases as well by use of phenotypic and molecular methods. The Vitek 2 system seems to represent an accurate and acceptable mean for performing characterization/ identification of many bacterial species [5]; however, identification of VS strains seems challenging also for this system [6]. Sequencing of the 16S rRNA gene is widely used in identification of bacteria and has revolutionized taxonomy over the last decades, though the discriminative power may call for additional molecular examinations when trying to separate VS strains and other closely related bacteria from each other [6, 7]. However, phylogenetic analysis based on 16S rRNA gene sequences of type strains of VS reveal a clustering pattern that reflect their pathogenic potential and ecological preferences [8].
We therefore found it interesting to characterize a large collection of VS strains obtained from blood cultures from a well defined group of infective endocarditis patients and a second group of strains from blood cultures assumed to be contaminants. Vitek 2 examination and partial sequencing of the 16S rRNA gene (a 526 basepair stretch) and subsequent BLAST examination were applied in order to compare and describe the methods and groups of strains, respectively. Materials and methods:
MATERIALS AND METHODS
Bacterial Strains
All strains of streptococci, Streptococcus-like bacteria, and Enterococcus faecalis (it was the only Enterococcus species which had been isolated from endocarditis patients) isolated at the Department of Clinical Microbiology at Herlev University Hospital in the period of January 1st. 2002-31 st. December 2006, from patients with definite infective endocarditis according to the modified Duke criteria [9] were included. In addition, VS strains from the same period, which were evaluated as blood culture contaminants, were included. Criteria for blood culture contamination were, if 1) only one or two of four blood culture bottles yielded growth within a given blood culture set and temperature, leucocyte(s) count(s) and C-reactive protein values of the patients were all within normal ranges, or 2) in the presence of another significant infection other than endocarditis with VS or another bacterial infection. All data were extracted from the laboratory information system (LIS) (AdBakt, Autonik, Sweden). Streptococcal isolates judged as contaminations were initially seldom attempted identified to the species level. All bacterial strains, including streptococcal isolates judged as contaminations were kept frozen at (-80ºC). The strains were taken from frozen, inoculated on 5% Danish Blood Agar and incubated at 35º C in ambient atmosphere supplemented with 5% CO2.
Phenotypic Identification
The Vitek 2 (BioMeriux) is an automated system for the species identification of bacteria [10]. After 6-8 hours of incubation, reactions were read automatically. A colorimetric GP card for the identification of Gram-positive cocci with 43 reactions [6] was used for identification of all isolates. When supplementary tests were recommended by the system, they were done, except for β-N-Acetyl-Galactosaminidase.
Partial 16S rRNA Gene Sequence Analysis
Partial 16S rRNA gene sequencing and subsequent BLAST examination was performed as described previously [7]. Briefly, DNA was released by heating isolated bacteria at 95°C for 5 min. PCR amplification of part of the 16S rRNA gene (526 base pairs stretch) was performed using the primers BSF-8 and BSR-534. All edited sequences were compared to deposited sequences in the GeneBank with a standard nucleotide- nucleotide BLAST approach (see Table 1). Blast files were stored electronically and later evaluated.
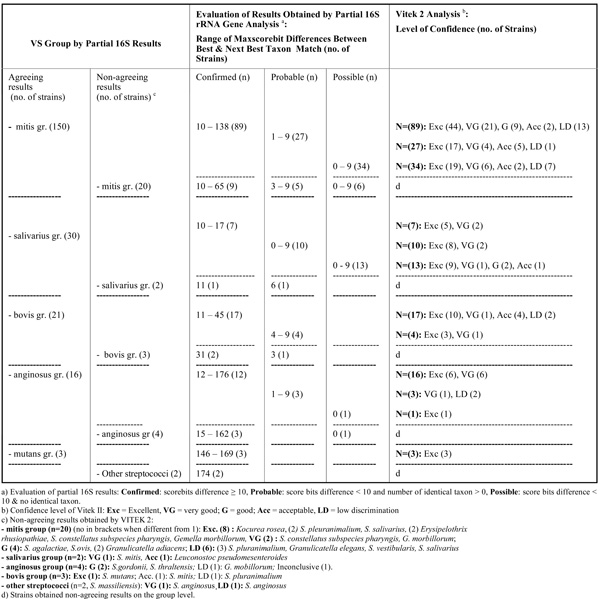
Results Obtained by Partial 16S rRNA Gene Sequence Analysis and Vitek 2 GP Card Testing for 251 VS Strains with Respect to Allocation the Same VS Groups 220 of 251 Strains Obtained Agreeing Results and 31 of 251 Strains Non-Agreeing Results
 |
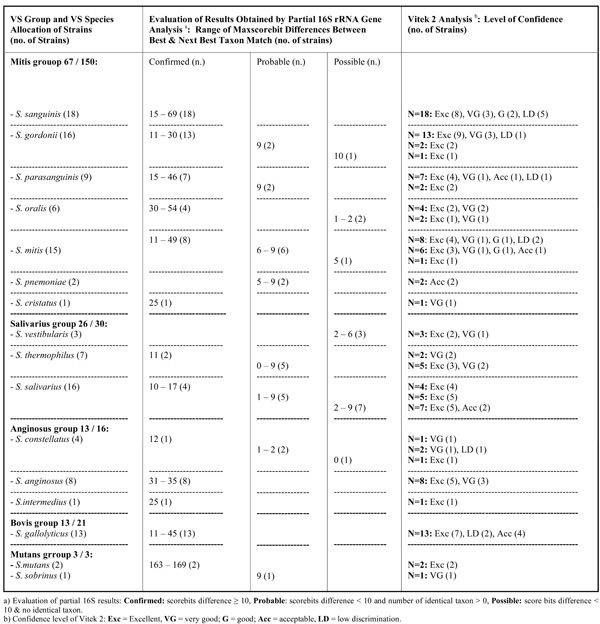
Results Obtained by Partial 16S rRNA Gene Sequence Analysis and Vitek 2 Testing for VS Strains Being Allocated to the Same VS Species by Both Methods (n=122)
 |
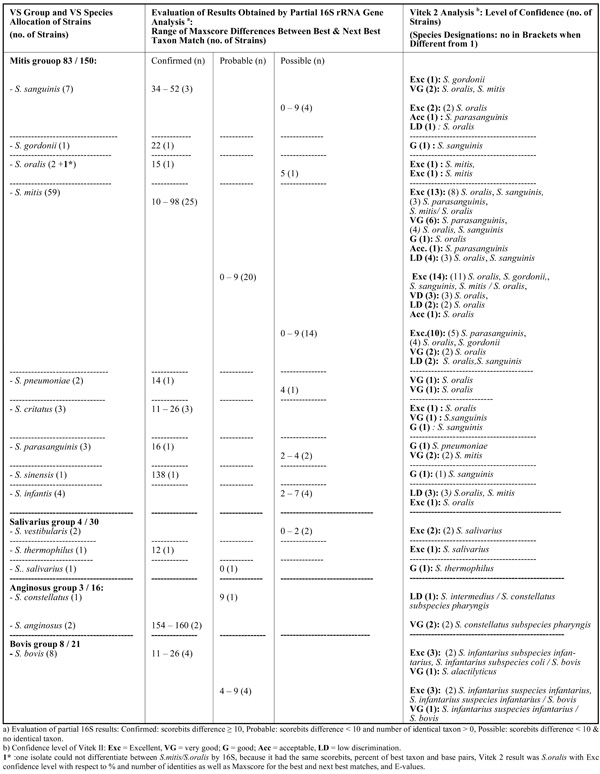
Results Obtained by Partial 16S rRNA Gene Sequence Analysis and Vitek 2 GP Card Testing for VS Strains not Allocated to the Same VS Species by Both Methods (n=98)
 |
RESULTS
In total 330 strains were included; 149 strains were from patients with definite infective endocarditis, and 181 strains from the same period, which had been evaluated as blood culture contaminants. Of the 330 strains, based on partial 16S rRNA gene sequencing results, 251 were VS strains, 10 were pyogenic streptococcal strains, 54 were E. faecalis strains and 15 strains belonged to a miscellaneous group of catalase-negative Gram-positive cocci.
In Table 1 results obtained by the two methods for VS strains with respect to allocation to the same VS groups are given. Of the 251 VS strains, 220 (87,6%) and 31 (12,3%), respectively, obtained agreeing and non-agreeing results on the group level. Within the mitis group of VS, 150 strains agreeingly were allocated to the same group, of which 89 strains achieved confirmed level by partial 16S rRNA gene sequence analysis with a range of Maxscore differences from 10 to 138 to the next best taxon match; most of the strains achieved excellent level of identification in the Vitek 2 system. Twenty of the 31 strains with non-agreeing results at the group level belonged to the mitis group of VS, judged by 16S rRNA gene sequence analysis. For the other VS groups mostly agreeing results were obtained.
Among the 220 VS strains allocated to the same VS group by both methods, non-agreeing species identifications mostly occurred among strains in the contaminant group (mitis group 66 / 83, salivarius group 3 / 4, bovis group 5 / 8, other streptococci 2 / 2 strains), while for endocarditis strains notably fewer disagreeing results were noticed (mitis group 17 / 83, salivarius group 1 / 4, bovis group 3 / 8). In Table 2a (n= 122) and 2b (n=98), data are given for strains obtaining and not obtaining, respectively, allocation to the same VS species by both methods. Only 67 of 150 mitis group strains obtained agreeing species identifications by both methods (Table 2a). Most VS strains belonging to the groups of salivarius, anginosus, bovis, and mutans obtained agreeing species identifications with the two methods, while this only was the case for 13 of the 21 bovis strains. Strains belonging to species in the mitis group had the highest number of disagreeing results being for 55,3% of strains (n = 83 /150) (Table 2b); respectively, 59, 7, 4, 3, 3, 3, 2, 1 and 1 strain(s) of the following species (when taking starting point in the partial 16S rRNA gene sequence analysis results): S. mitis, S. sanguinis, S. infantis, S. parasanguinis, S. cristatus, S. oralis, S. pneumoniae, S.gordonii and S. sinensis). Among the non-agreeing S. mitis strains, 25 of 59 strains obtained species confirming results (score bits difference 10 to 98) by partial 16S rRNA gene sequencing/BLAST examination.
All ten pyogenic streptococcal strains achieved agreeing results by the two methods. Five strains had a Maxscore difference between best and next best taxon match of 82-107 and 5 strains a difference of 0-25; by Vitek 2 8, 1 and 1 strain(s), respectively, were recorded as Excellent (Exc), Very Good (VG) and Low Discrimination (LD) level identifications. The following haemolytic species were identified: S agalactiae (n = 4), S. dysgalactiae. ssp. equisimilis (n = 4), S. pyogenes (n = 1), and S. equi ssp. zooepidemicus (n = 1). All 54 E. faecalis strains had Maxscore differences between best and next best taxon match of 121-171 with excellent identifications by Vitek 2.
In the miscellaneous group of strains (n=15) agreeing genus identifications were obtained for 9 strains of which 7 strains obtained confirmed partial 16S rRNA gene sequencing identifications (range of Maxscore difference: 12-85) and 2 possible identifications [Granulicatella species strains (n=2) with Maxscore differences of 5]. The 9 strains belonged to the following genera: Globicatella (n=3), Granulicatella (n=4) and Gemella (n=2) and agreeing species identifications were obtained for 5 of these strains (respectively Globicatella sanguinis (n=2), Granulicatella adiacens (n=1), Gemella morbillorum (n=1) and Gemella haemolysans (n=1)). Six strains with confirmed partial 16S rRNA gene sequencing identifications (range of Maxscore difference: 12-155) obtained non agreeing Vitek 2 identifications (partial 16S rRNA gene sequencing/Vitek 2 identifications: G. sanguinis/S. pneumoniae; G. morbillorum (n=2)/Gardnerella vaginalis; G. adiacens/S. alactolyticus; Facklamia languida/Kocurea rosea).
DISCUSSION
In this study we compared the results obtained by Vitek 2, using the colorimetric GP card for the identification of Gram-positive cocci with 43 phenotypic reactions, and by partial sequencing of the 16S rRNA gene (a 526 basepair stretch) and subsequent BLAST examination on a large number of strains including VS strains (n = 251 isolates), haemolytic streptococci (n =10), E. faecalis (n = 54) and Streptococcus-like bacteria (n = 15). The strains were grown from blood-cultures from patients with verified infective endocarditis as well as from patients without infective endocarditis, where blood-culture contamination was suspected. Notably, among VS strains allocated to the same VS group, most disagreeing results, with respect to allocation to the same VS species, occurred among assumed contaminant strains. These findings are in agreement with the findings by Haanperä et al. [6], who used a pyrosequencing method for the identification of streptococcal species based on two variable regions of the 16S rRNA gene. Identification of members of the S. mitis and S. sanguinis groups proved difficult for both the pyrosequencing method and the Vitek 2 system. Furthermore, the pyrosequencing analysis revealed great sequence variation, since only 43 (32.3%) of 133 strains analyzed by pyrosequencing had sequences identical to a type strain. The variation was highest in the pharyngeal strains, slightly lower in the blood culture strains, and nonexistent among invasive pneumococcal isolates (n = 17), that all had the Streptococcus pneumoniae type strain sequence. Whether this reflects a difference among disease provoking and contaminating VS strains seems worth to explore in more detail.
VS groups may often be separated from other groups of Gram-positive cocci and from each other on basic phenotypic reactions [2, 11, 12]. They are alpha- or gamma-hemolytic, form chains, are catalase negative, produces leucine aminopeptidase, but are without pyrrolidonyl aminopeptidase activity. For VS group identification, results obtained for arginine decarboxylase production, esculin hydrolysis, Voges-Proskauer reaction, urease production and acid production from mannitol and sorbitol are of great help. Especially the high no. of strains from the mitis group species can be problematic [12]. Identification to the species level therefore in all times has been a challenge. Many attempts to solve species identification within VS strains have been made over time, including kit-based systems. Funke and Funke-Kissling [13] was the first to publish an evaluation of the new Vitek 2 colorimetric GP card and found all of the 18 alpha-hemolytic streptococcal species examined to be correctly identified. Haanperä et al. [6] used a pyrosequencing method (see above). Almost all studied streptococcal species (n = 51) represented by their type strains were differentiated, except some closely related species of the S. bovis or S. salivarius group. Pyrosequencing results of alpha-hemolytic strains from blood (n = 99) or from the normal pharyngeal microbiota (n = 25) were compared to the results obtained by the Vitek 2 with the colorimetric GP card. The results of the two methods did not completely agree, but 93 (75.0%) of the isolates were assigned to the same streptococcal group by both methods and 57 (46.0%) reached consistent results at the species level. These data are in line with our findings, that among VS strains, respectively, 220 (87,6%) and 31 (12,3%) obtained agreeing and non-agreeing identifications with the two methods with respect to allocation to the same VS group and of strains allocated to the same VS group, 55,5 % (122 of 220 strains) were allocated to identical species by the two methods. These obtained data, however, covers a continuum of certainty, as searched illustrated by dividing molecular data into confirmed, probable and possible. Though most strains typically are given in the confirmed column, the identification challenges within especially the mitis group is clearly illustrated by the great no. of strains given in the probable and possible columns. Likewise, the spread in identification level obtained by Vitek 2 illustrates this uncertainty. In the work by Haanperä et al. [6], 10 strains remained unidentified by Vitek 2, and 4 strains could not be assigned to any streptococcal group by pyrosequencing. Most of discrepant results were found in the mitis and sanguinis groups, being in agreement with our results. Inclusion of tests for acid production from inulin and production of dextran will probably be able to contribute significantly to discrimination within the mitis group. For the other frequently encountered VS groups, the anginosus groups, salivarius and bovis groups, there seems to be less discriminatory problems with the tests included in the system.
For correct molecular species identification of VS strains, sequencing of other genes or application of other methods therefore seems desirable. Simmons et al. [14] examined 94 VS strains from 94 patients with definite endocarditis to evaluate the phylogenetic relationships of VS with 16S rRNA, tuf, and rpoB gene targets. The used 16S rRNA primer pairs were 5F and 534R, which is very similar to the bp stretch used in this study. Overall, VS isolates demonstrated a high degree of variability for all three targets, which was not found a surprising observation since transfer of genetic material among streptococci has been well described [8, 14, 15]. In their study, classifications within groups were not always predictable or correlated with phenotype or phylogeny; an observation also noted by Hoshino et al. for isolates from patients with bacteremia and meningitis [4]. Kilian et al. [8] have looked at the evolution of S. pneumoniae and its close commensal relatives. A population genetic analysis of 118 strains tentatively assigned to S. pneumoniae, S. mitis, S. oralis, and S. infantis, aligning the sequences of the four housekeeping genes, ddl, gdh, rpoB, and sodA, plus eight sets of sequences extracted from S. pneumoniae genomes revealed, a remarkable sequence polymorphism and that S. pneumoniae is one of several hundred evolutionary lineages forming a cluster separate from S. oralis and S. infantis. The remaining lineages of this distinct cluster are S. mitis strains (commensals).
Also rnpB, sodA, and/or 16S-23S rRNA spacer targets have been used for characterization of strains illustrating the mentioned complexity [16-18]. For daily routine characterization and identification of VS the use of a combination of sequence analysis of the ITS region and of the partial gdh gene have proven promising [19].
For practical use, pyogenic streptococci are characterized by phenotypic and serological characteristics [2]. However, if reactions are found inconclusive, Vitek 2 can be very helpful or sequencing parts of the 16S rRNA gene.
All 54 E. faecalis strains seemed well identified by both methods.
The Streptococcus-like bacteria (Globicatella, Granulicatella, Gemella, Facklamia and Aerococcus species) identified in our study, are rare but well described aetiologies of infective endocarditis [11, 20-28]. Most are well characterized by molecular methods [11, 21, 23, 29-32], but hardly included in the databases used for kit-based/automated phenotypic identification systems. Therefore, phenotypic characterization not seldomly is a major laboratory challenge.
CONCLUSION
In conclusion, the present study shows the usefulness of the Vitek 2 GP card and partial 16S rRNA gene sequence analysis for identification of VS strains to the group level, but also the problems occurring when searching to reach a species identification, which seems most problematic with the mitis group of species. Interestingly, among VS strains non-agreeing species identification mostly occurred among strains in the contaminant group, while for endocarditis strains notably fewer disagreeing results were observed.

