All published articles of this journal are available on ScienceDirect.

Glycolipid-Dependent, Protease Sensitive Internalization of Pseudomonas aeruginosa Into Cultured Human Respiratory Epithelial Cells
Abstract
Internalization of PAK strain Pseudomonas aeruginosa into human respiratory epithelial cell lines and HeLa cervical cancer cells in vitro was readily demonstrable via a gentamycin protection assay. Depletion of target cell glycosphingolipids (GSLs) using a glucosyl ceramide synthase inhibitor, P4, completely prevented P. aeruginosa internalization. In contrast, P4 treatment had no effect on the internalization of Salmonella typhimurium into HeLa cells. Internalized P. aeruginosa were within membrane vacuoles, often containing microvesicles, between the bacterium and the limiting membrane. P. aeruginosa internalization was markedly enhanced by target cell pretreatment with the exogenous GSL, deacetyl gangliotetraosyl ceramide (Gg4). Gg4 binds the lipid raft marker, GM1 ganglioside. Target cell pretreatment with TLCK, but not other (serine) protease inhibitors, prevented both P. aeruginosa host cell binding and internalization. NFkB inhibition also prevented internalization. A GSL-containing lipid-raft model of P. aeruginosa host cell binding/internalization is proposed
INTRODUCTION
Pseudomonas aeruginosa is an opportunistic pathogen primarily associated with the lung pathology and morbidity of cystic fibrosis patients [1]. It is highly motile due to flagella and type 4 pili (T4P)-mediated twitching [2]. T4P are also involved in attachment of the organism to both target cells and inert surfaces [3]. The organism is also prone to form T4P dependent biofilms [4], sessile protected microcolonies, at the site of infection making antibiotic management difficult [5]. P. aeruginosa expresses a broad spectrum of pathological virulence factors responsible for disease [6].
The organism is largely an extracellular pathogen, however, internalization of P. aeruginosa into target cells has been reported [7, 8]. This internalization process has been proposed to be, in part, responsible for the normal elimination of the organism by induction of host cell apoptosis and macrophage clearance, a process proposed deficient in cystic fibrosis cells [9].
The neutral glycosphingolipid (GSL) gangliotetraosyl ceramide, Gg4 (also referred to as asialoGM1) has been reported to bind many pathogenic bacteria including P. aeruginosa [10, 11]. Gg4 has been proposed to provide the respiratory epithelial cell receptor for P. aeruginosa infection [12-16]. Gg4 expression levels have been shown to be increased in epithelial cells derived from cystic fibrosis patients, consistent with their enhanced susceptibility to infection [17]. We have verified increased Gg4 expression in a CF-derived respiratory cell line [18]. However, we also demonstrated that P. aeruginosa does not bind to Gg4 in solid phase binding assays or when expressed within the plasma membrane of target host cells [18]. We did nevertheless, find that T4P sheared from the P. aeruginosa organism could specifically bind Gg4 (and Gg3) in an in vitro receptor ELISA. We therefore, have continued to explore the potential role of GSLs in P.aeruginosa pathology.
Internalization of the standard PAK P.aeruginosa laboratory strain into eukaryotic cells is src kinase dependent [19-21] and is modulated by CFTR and caveolin1 [8]. We now show the P. aeruginosa internalization pathway is a GSL-dependent process. Prevention of binding and internalization by protease inhibition suggests a novel src kinase dependent P.aeruginosa host cell interaction mechanism.
MATERIALS AND METHODS
Bacterial Strains and Cultured Cells
P. aeruginosa strains PAK [22], PAK-np (kindly provided by Dr L. Burrows, McMaster University), and Salmonella typhimurium SL 1344 were maintained in glycerol at -80 ºC. Bacteria were grown on Luria-Bertani (LB) agar plates, over night at 37 ºC prior to epithelial cell infection. IB3-1 (derived from a CF patient with ΔF508/W1282X mutant CFTR) and S9 (transfected IB3-1 cells transfected with full length wildtype CFTR) brochial epithelial cells were grown as previously [18]. HeLa cervical epithelial carcinoma cells were from ATCC and maintained as described [23].
GSL Depletion
P4 (1-phenyl-2-palmitoylamino-3-pyrrolidino-1-propanol) was a generous gift from Dr. J. Shayman, University of Michigan. It is an inhibitor of glucosylceramide synthase, the enzyme catalyzing the first glycosylation step in the synthesis of glucosylceramide-based glycosphingolipids [24] to prevent the synthesis of most gangliosides and neutral GSLs. IB3-1-1, S9 and HeLa cells were maintained in medium containing 1µM P4 reconstituted in DMSO, for at least 10 days as previously [18]. To test the effect of selective neutral GSL depletion on P. aeruginosa internalization, cells were treated with 8µM cyclosporine A [25] for a minimum of 3 days prior to confluency and bacterial internalization assay.
Synthesis of Deacetyl Gangliotetraosyl Ceramide
A solution of gangliotetraosyl ceramide (Gg4, 2 mgs, 1.5 µmol, weighted average molecular weight, 1291.8 gmol-1, prepared from acid hydrolysis of GM1 ganglioside [26]), was portioned into a reaction tube and dried under a flow of nitrogen. Sample was kept under vacuum in a desiccator containing phosphorus pentoxide for 6 hours. The dry sample was dissolved in methanolic NaOH (2 mLs of 1 M NaOH in dry MeOH), sonicated and heated at 72 to 74 C for 2 hours. The reaction mixture was cooled to room temperature, neutralized with HCl (6 M HCl) and most of the methanol was removed under vacuum to yield a syrupy material. This material was dissolved in water (15 mLs), desalted on a C-18 reverse phase column and the methanolic fraction was dried. Sample was then dissolved in CHCl3: MeOH: H2O; 90: 10: 0.5 (3 mLs) and loaded on to a silica column (silica: 35-70 micrones, suspended in CHCl3: MeOH; 98: 2, bed volume: 1.5 cm o. d., 2 cm height) column and eluted with the following solvents; C: M: H2O; 90:15:1 (1, 15 mL fraction), C: M: H2O; 80: 20: 2 (6, 2 mL fractions), C: M: H2O; 65: 25: 4 (until product was eluted).
TLC Overlay to Detect GSL-GSL Binding
2µg GSL were spotted on TLC. Plates were blocked for 1hr at room temp. with1%BSA in PBS and washed 3x in PBS. The plates were incubated with 10µg/mL Gg4 in RPMI 1640 containing 10% FBS for 30 mins at 37oC. The plates were washed and incubated with Mab antiGg4 for 2 hr at room temp., washed, and incubated with HRP conjugated anti mouse antibody for 1hr. After washing, bound antiGg4 was detected using 4-chloronaphthol.
Gentamycin Resistance Assay for Quantification of Internalized Bacteria
IB3-1 and S9 cells P4 treated and untreated were grown in LHC-8 media. HeLa cells were grown in DMEM media. All cell lines were grown in 6 or 12 well plates at 37ºC to reach confluency for internalization assay. Bacteria were grown on LB plates over night, scraped from the plate and washed in PBS by centrifugation at 3000 r.p.m. for 5 minutes. They were resuspended in RPMI-160 with 10% fetal bovine serum (FBS) supplemented with 1% Hepes buffer to OD of 0.07-0.09 (~ 108 bacteria ml-1). Salmonella typhimurium was grown in 2 ml LB broth with 50 µg/ml of gentamycin at 37 ºC, over night to remove any hypersensitive organisms, and subcultured (300 µl was added to 10 ml of LB) for a further 3 hours at 37 ºC. The culture was then centrifuged at 10000g for 2 minutes, resuspended in PBS supplied with Mg2+ and Ca2+ and adjusted to an OD of 0.1 prior to infection. Bacterial cultures were confirmed to be 100% susceptible to 400µg/mL gentamycin. Cells were infected with bacteria (M.O.I of ~100) for 30 minutes. This time was the maximum under which P.aeruginosa host cell binding was T4P-dependent [18]. Unbound bacteria were washed off three times with PBS. The remaining extracellular bacteria were killed with 400 µg.ml-1 gentamycin in RPMI 1640, for 2 hours at 37 ºC. Cells from at least three wells were washed and lysed with 0.5% Triton X-100 (Sigma) in PBS for 15 minutes at room temperature. Aliquots of cell lysates were serially diluted and plated on LB plates. The plates were incubated over night at 37 ºC and the colonies were counted the next day.
To test the effect of soluble GSL analogues or inhibitors on P. aeruginosa internalization, epithelial cells were grown in 96 well plates just to confluency and preincubated with varying concentrations of the compounds for 30 minutes. Cells were washed with medium 3x prior to P. aeruginosa infection and internalization.
Statistical Analysis
Data for gentamycin protection assays are reported as a mean of at least three experiments with error bars representing standard deviation. Significance was calculated using a two-tailed t-test with significance defined and a p value of less than 0.05.
Electron Microscopy TEM
For transmission electron microscopy, S9, IB3-1-1 and P4 treated S9 cells were grown in 10-cm-diameter tissue culture dishes until confluent. The cells were then infected with P. aeruginosa PAK, as described above. After washing six times in PBS to remove nonadherent bacteria, the monolayers were fixed with 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, for 10 minutes at room temperature. Cells were scraped from the tissue culture dishes and centrifuged at 600 rpm in the buffered fixative. The cell pellets were next postfixed in 1% aqueous osmium tetroxide for 1 h. Dehydration was then performed in graded acetone, followed by embedding in epoxy resin. Osmium fixation, dehydration, and embedding were conducted in a Pelco Biowave microwave oven (Pelco International, Redding, CA). One-micrometer-thick sections were stained with toluidine blue, and ultrathin sections were stained with uranyl acetate and lead citrate. Transmission electron microscopy examination was performed using a JEM 1011 (Joel USA Corp., Peabody, MA) transmission electron microscope with a CCD camera system (AMT, USA.) attached.
RESULTS
GSL Dependency of P. aeruginosa Invasion
P. aeruginosa internalization into IB3-1 and S9 cells could be readily detected by protection from gentamycin. There was no significant difference between internalization into these cell lines (Fig. 1). P. aeruginosa internalization into HeLa cells was slightly less effective. Although we have shown that GSL depletion of target cells has no effect on initial P. aeruginosa adhesion [18], depletion of neutral and acidic host cell GSLs by P4 had a major inhibitory effect on P. aeruginosa internalization in S9, IB3-1 and HeLa cells (Fig. 1a). In contrast, selective inhibition of neutral GSL biosynthesis with cyclosporin had little inhibitory effect (Fig. 1b), indicating the acidic ganglioside GSL fraction was of primary importance. Internalization was pili dependent since the T4P mutant P. aeruginosa strain did not internalize (Fig. 1c). However, this may result from the lack of cell attachment seen for this mutant [18].
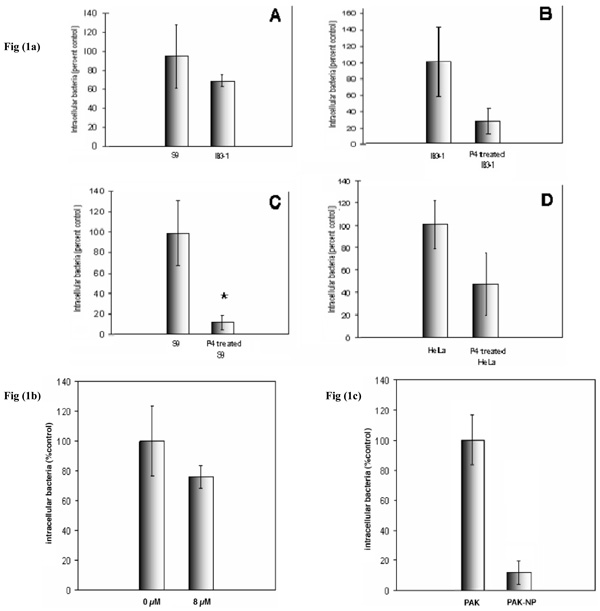
P. aeruginosa internalization into airway epithelial cells. a)effect of total GSL depletion. IB3-1 and S9 cells were treated ± P4, infected with P. aeruginosa at an M.O.I. of 100 for 30 minutes and internalization was quantified by gentamycin protection assay. Panels show comparison of P. aeruginosa internalization by IB3-1 and S9 cells (A), and internalization by IB3-1 (B) and S9 (C), before and after P4 treatment. P. aeruginosa internalization by HeLa cells before and after P4 treatment was also measured (D). P. aeruginosa internalization into control S9, IB3-1 and HeLa cells were ~105, 106 and 3x104P. aeruginosa /107 cells respectively. Data represent means ± SD of three experiments. b) effect of neutral GSL depletion. S9 cells were treated with cyclosporin A for 72 hours prior to internalization assay. CsA selectively inhibits neutral GSL biosynthesis. Cells were infected with an M.O.I. of 100 for thirty minutes. The concentration of cyclosporine A added to the growth medium in µM is shown. Data represent means ± SD of three experiments. c) effect of T4P depletion S9 cells were infected with PAK or its mutant non-piliated strain PAK-np at an M.O. I. of 100 for 30minutes. Data represent mean ± SD of three individual experiments. Non-piliated organisms are not internalized. * indicates p<0.05.
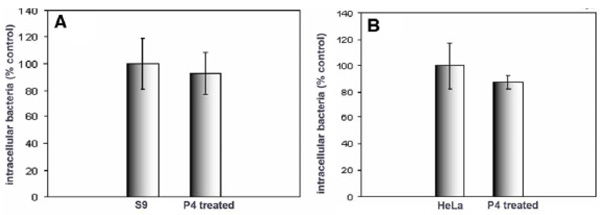
Effect of GSL depletion on Salmonella typhimurium internalization by S9 and HeLa cells. S9 (A) and HeLa (B) cells were treated with P4, and infected with Salmonella for 30 minutes prior to quantification of internalization. Data represent means ± SD of three experiments. Internalization was unaffected by P4 treatment

Electron microscopy of internalized P. aeruginosa. a) S9 cells ±P4 pretreatment were infected with PAK bacteria and after 30 mins, were fixed and processed for EM. a) comparison of untreated (upper) and P4 treated cells (lower). Internalized P. aeruginosa are arrowed. Bar= 4µM. b) Intracellular P. aeruginosa within host cell vesicles A-F. In panels A,B,C the S9 cell membrane is uppermost. Bar= A-E 500nm F,100nm. Organisms are internalized for the most part, into individual membrane vesicles. The vesicle membrane is closely apposed to the bacterium for organisms proximal to the cell surface (A,B,C). For more distal vesicles, a space between these membranes is apparent (B,C). This space contains microvesicles (D-F) associated with both the host (D,E) and bacterial (E,F) membranes. No overt morphological distinction was seen for the few P. aeruginosa containing vesicles in P4 treated cells.
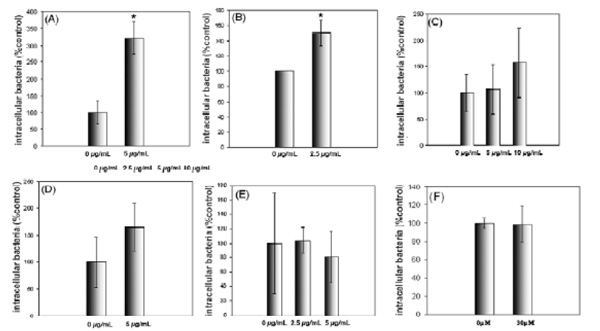
Exogenous glycosphingolipids can increase P. aeruginosa uptake by airway epithelial cells. Cells were incubated with deacetylGg4 for 30 minutes prior to internalization assay, they were infected at an M.O.I of 100. Internalized bacteria were quantified by gentamycin protection assay. A shows the results of internalization by S9, B shows internalization by IB3-1, after pretreatment with deacetylGg4. C shows internalization by IB3-1, after pretreatment with Gg4, D shows internalization by P4 treated IB3-1 cells after pretreatment with deacetylGg4, E shows the change in P. aeruginosa internalization into IB3-1 cells after pretreatment with exogenous deacetylGb4 and F shows the effect of BAPTA on P. aeruginosa internalization into IB3-1 cells. P. aeruginosa internalization is significantly enhanced by pretreatment with deacetylGg4 but not deacetylGb4. Native Gg4 is less effective and deacetylGg4 stimulation is not significant for P4 treated cells. Calcium chelation by BAPTA does not affect internalization indicating no role for GSL regulated purine receptors. The concentration of exogenous GSL analogues in µg/mL are shown. Data represent means ± SD of three experiments. * indicates p<0.05.

GSL binding of deacylGg4. GSLs were applied to tlc plates dried and detected by orcinol spray for carbohydrate (upper panels). A separate series of GSL dot blots was incubated with deacylGg4 and any bound Gg4 detected by immunostaining with Mab antiGg4 (middle panels). a) GM3 ganglioside, b) GM1 ganglioside, c) Gg3, d) Gb4, e) LacCer. No reaction was seen in the absence of antiGg4 or without incubation with deacylGg4. Bound deacylGg4 detected for each GSL was quantitated using Image J (lower panel). DeacylGg4 bound LacCer>Gg3>GM1.

TLCK prevents P. aeruginosa internalization. A) S9 cells were pretreated with 50µM TLCK for 1hr at 37°C prior to assay of P. aeruginosa internalization. The effect was compared to that of P4 GSL inhibition. B) the effect of TLCK on P. aeruginosa colonies for S9 cells with (internalized bacteria only) and without (internalized+host cell attached bacteria) gentamycin was measured. C) S9 cells (lane 1) were pretreated with P4 (lane 2) or 100µM AEBSF for 30mins. D) S9 cells (lane 1) were pretreated with P4 (lane 2), 10µM PPM-18 for 1hr (lane 3) or 50µM suramin overnight (lane 4). Data represent means ± SD of three experiments. * indicates p<0.05.
We compared the effect of GSL depletion on the internalization of P. aeruginosa and Salmonella typhimurium into the same cells viz HeLa cells, commonly used in the study of Salmonella internalization, and S9 cells. Salmonella typhimurium is an obligate intracellular pathogen and the mechanism of host cell invasion is well characterized, involving a type III secretion system [27, 28]. Although interaction between Salmonella components and host cell GSLs have been reported [29], no role in internalization has been implied. In contrast to P. aeruginosa, Salmonella internalization into either S9 or HeLa cells was unaffected by GSL depletion (Fig. 2).
P. aeruginosa Containing Vacuoles
Electron microscopy showed that the GSL-dependent internalized P. aeruginosa were contained within tightly apposed membrane vacuoles (Fig. 3a, 3b). In cross section, only a single organism per vacuole was seen (Fig. 3bA). Occasional longitudinal sections showed two, head to head organisms. Organisms near the cell periphery tended to show close apposition of bacterial and host vacuole membrane. In the more central vacuoles in the cell, a significant space between these membrane was observed and this space contained very small bilayer vesicles (Fig. 3b E,F). A few such vesicles could be distinguished between the bacterium and host membrane even in the tightly enveloped vacuole.
GSL Stimulation of P. aeruginosa Invasion
Although P. aeruginosa do not bind GSLs on target cells, we established that the type IV pili (T4P) when released from the organism, can in fact, bind Gg4 [18]. Since the pili are lost from internalized P. aeruginosa, we questioned whether the GSL binding T4P phenotype played any role in GSL dependent P. aeruginosa invasion. As a first step, potentially competitive exogenous GSLs were tested. Pretreatment of S9 or IB3-1 cells with purified deacetylGg4 resulted in a remarkable stimulation of P. aeruginosa internalization (Fig. 4). Gg4 had a similar, but less potent stimulatory effect. A control GSL analogue deacetylGb4, had no effect. When cells were pretreated with P4 to deplete endogenous GSLs, deacetylGg4 stimulation less, suggesting endogenous GSLs play a role in the stimulation. To determine whether the known linkage between Gg4 and calcium-dependent purine nucleotide receptor signaling [30, 31] could play a role, the effect of the standard calcium chelator, BAPTA [32] was assessed. This did not reduce P. aeruginosa internalization, suggesting that the BAPTA-sensitive purine receptor signaling pathway is not involved.
DeacetylGg4 Binds to GM1 Ganglioside
Since GSL depletion was found to reduce deacetylGg4 stimulated P. aeruginosa internalization, we considered whether endogenous GSLs could play a role in this process. We assessed whether a carbohydrate-carbohydrate interaction might be important by determining the direct interaction of deacetylGg4 with a panel of purified GSLs by using a TLC overlay immunodetection procedure (Fig. 5). We found significant binding to GM1 ganglioside, Gg3 and lactosyl ceramide (LacCer). No binding to Gb4 was observed. The lack of binding to GM3 as compared to strong binding to LacCer, indicates charge plays no role, or even inhibits, deacetylGg4–GSL binding.
Protease Inhibition Prevents P. aeruginosa Epithelial Cell Invasion
S9 represents wildtype respiratory epithelial cells, strongly protected by GSL depletion. Brief pretreatment of the S9 cells with the trypsin inhibitor, TLCK prevented P. aeruginosa internalization as effectively as GSL depletion (Fig. 6). Elimination of the antibiotic treatment to assess cell bound, in addition to internalized organisms (Fig. 6B) shows that TLCK reduces P. aeruginosa cell binding, in addition to internalization. We estimate 1 organism is internalized/100 untreated cells and previous assessment of binding [18] shows ~4 organisms bound/cell, giving an internalization efficiency of ~0.25%. Thus TLCK treatment of target cells prevents >99% P. aeruginosa cell binding. The inhibitory effect was not a consequence of trypsin inhibition since the trypsin inhibitor, AEBSF was without effect.
TLCK also is active against some serine proteases such as thrombin and plasmin and some cysteine proteases such as papain. In addition, TLCK is a selective inhibitor of NFκB [33]. In preliminary studies, we established that the NFκB inhibitor, PPM-18 [34] also prevented P. aeruginosa internalization (Fig. 6C panel A), suggesting that this transcription factor could be involved in the GSL dependent P. aeruginosa entry process. Suramin, an inhibitor of tyrosine phosphatases [35], CFTR [36] and serine proteases [37], had no significant effect on P. aeruginosa internalization (Fig. 6C panel B).
DISCUSSION
GSL Dependent Internalization
We have shown a GSL-dependent pathway for the selective internalization of Pseudomonas aeruginosa into human epithelial cells. Treatment of target cells with P4 under conditions to completely deplete GSLs [18] prevents internalization. This is not a general prevention of bacterial invasion since Salmonella internalization is unaffected. P. aeruginosa without T4P were not internalized but this mutant is defective in target cell binding [18] which is likely a crucial step for internalization. The intracellular vacuoles containing P. aeruginosa are morphologically consistent with previous studies on P. aeruginosa invasion [38] in which the vacuoles were defined as endosomes fused with secondary lysosomes [39]. PLC-dependent vaculolar membrane destruction and free cytosolic bacteria have been observed within one hour [39]. While free cytosolic bacteria were not found in our short-term studies, the small intravacuolar vesicles observed for the more distal organisms could represent an early stage of this escape process. P. aeruginosa produce such vesicles in culture and these contain virulence factors including PLC and proteases [40]. These vesicles are increased by gentamycin treatment, but gentamycin was not used in our EM studies. Moreover, bacteria more distant from the plasma membrane were associated with more vesicles than those more proximal to the cell surface, presumably more recently internalized, and a vesicle-containing space between the bacterial and host cell membranes becomes apparent. This would be consistent with intracellular induction of bacterially derived vesicle formation and the destruction of the host vacuolar membrane by the hydrolytic enzymes they contain.
GSLs and Src Kinase Rafts
P. aeruginosa host cell invasion is dependent on tyrosine phosphorylation and inhibition of lipid raft associated src kinases [19-21, 41] or the src-related Abl kinase [42] prevents internalization without affecting P. aeruginosa cell binding. Raft depletion per se, via cholesterol restriction, also prevents internalization [41, 43] and depletion of the cholesterol binding caveolin1 also restricts entry [8]. Src kinases are found associated with the cytosolic face of GSL enriched plasma membrane lipid microdomains or rafts [44]. Lipid rafts containing CFTR have been implicated in P. aeruginosa internalization [8, 45 , 46]. Gangliosides, particularly GM3 [47-50], have been found associated with Src kinases and have been shown to modulate Src kinase activity [51]. These GM3/src kinase containing domains have been termed a ‘glycosynapse’ [52]. GM3 can selectively recruit Csk, a negative regulator of src kinase, to such a glycosynapse [53]. Gangliosides can also modulate growth factor receptor kinase activity within lipid rafts [54, 55]. Thus, GSL modulation of raft kinase signaling may provide the basis of the GSL dependent P.aeruginosa internalization we have found.
Inhibition of glucosyl ceramide synthase prevents the synthesis of most GSLs(neutral and acidic) [24]. A few galactosyl ceramide-based GSLs (primarily galactosyl ceramide and sulfogalactosyl ceramide) remain unaffected. We have previously shown that inhibition of MDR1 by inhibitors such as CsA, results in the depletion of cell neutral GSLs without affecting the acidic ganglioside fraction [25, 56]. Our finding that P4 was more effective to prevent internalization than cyclosporin A, may therefore implicate target cell gangliosides in this process. GM1 is the major ganglioside of many cells and is used as the gold standard lipid raft marker in many studies [57]. GSL-enriched lipid rafts are involved in the internalization of many microbial pathogens [58] including P. aeruginosa [41 , 43]. Stimulation of P. aeruginosa internalization by pretreatment of target cells with Gg4, might indicate that raft GM1 mediates the GSL-dependent internalization pathway, if Gg4 was sialylated to form GM1. Gg4 has been shown to accumulate in CF cells [17, 59] and we have verified this for IB3-1 versus S9 cells [18]. The Gg4 accumulation in CF cells has been ascribed to a defect in sialylation [60] (which would reduce GM1 synthesis), induced by an altered Golgi pH in CF cells, resulting in the accumulation of Gg4 [61]. However, the common GM1 is GM1a in which the sialic acid is linked to the internal galactose. Gg4 (asialoGM1) is not the precursor of GM1a and therefore, lack of sialylation would not result in Gg4 accumulation. Thus, exogenously added (or endogenous) Gg4 would not be anabolized to generate GM1a ganglioside.
Ligation of Gg4 has been shown to mediate signal transduction through the purine nucleotide receptor [31] to activate Erk kinase and this has been shown to augment signaling through Toll-like receptor 5 [30]. However, this process of Erk activation has been shown to be strictly calcium dependent and we were unable to demonstrate any effect of calcium chelation in our studies, suggesting that Gg4/purine nucleotide receptor signaling is not central.
GSLs are poorly water-soluble and this hampers their use as pharmacological tools. We have designed a procedure for the generation of soluble GSL mimics which retain bioactivity by exchanging the GSL fatty acid for an adamantane frame [62]. The generation of such mimics for aminosugar containing GSLs is more complex since deacetylation of the amino sugar can be a confounding problem. We have developed procedures for the selective deacetylation/reacetylation of amino sugars and deacylation/reacylation of the sphingoid base [63]. In the present studies however, we found that deacetylation of the amino sugar of Gg4 alone was sufficient to engender significant water solubility, likely due to the generation of the charged amino function. DeacetylGg4 was more stimulatory for internalization than was Gg4 itself, and exogenous Gg4 has been previously reported to stimulate P. aeruginosa internalization [15]. Pretreatment of the bacteria, rather than target cells, with deacetylGg4 had no effect on subsequent target cell internalization. Thus, the effect is dependent on host cell factors. In the time frame of our experiments, exogenous deacetylGg4 could be reacetylated and incorporated as a plasma membrane component. IB3-1 cells contain more endogenous Gg4 than S9 cells and the surface expression of Gg4 in IB3-1 cells is much greater than S9 cells (barely detectable) [18]. Since IB3-1 cells were slightly less susceptible to P. aeruginosa internalization, a direct role for endogenous Gg4 is unlikely.
The role of cell internalization in clinical P. aeruginosa pathology, particularly in cystic fibrosis, remains controversial. Defective binding and internalization in CFTR deficient cells may reduce apoptosis to promote inflammation in CF [9]. However, P. aeruginosa host cell binding can be independent of CFTR surface expression [18, 64]. Despite the significantly increased Gg4 content of IB3-1 compared to S9 cells [18], GSL dependent P. aeruginosa internalization was not significantly different from S9 cells. Although our studies only define a virulence mechanism of P. aeruginosa, rather than, as yet, provide insight into the pathology of this infection in cystic fibrosis, inhibition of epithelial cell GSL synthesis (by an inhibitor of glucosyl ceramide synthase similar in action to P4) results in the loss of P. aeruginosa induced inflammation [65].
Our studies infer a greater importance of gangliosides in P. aeruginosa internalization and inhibition of endogenous GSL synthesis compromised the stimulatory effect of exogenous deacetylGg4. This would be consistent with a role for endogenous GM3 ganglioside in src signaling and P. aeruginosa internalization. Src family kinases bind the GSLs, sulfogalactosyl ceramide and GM3 ganglioside in vitro [66]. GM3/src provides a potential target for deacetylGg4 stimulation of P. aeruginosa internalization. However, our in vitro binding assay showed deacetylGg4 bound to several GSLs, but not GM3 ganglioside. (Thus, deacetylGg4-GSL binding is not charge dependent.) This previously unreported [67] carbohydrate-carbohydrate interaction with GM1, could implicate GM1 in the deacetylGg4 stimulation mechanism. Src kinases are associated with cholesterol/GSL enriched lipid rafts, marked by the presence of GM1 ganglioside. Src kinase activity regulates caveolin1-mediated caveolar endocytosis [68] and decreasing caveolin-1 decreases P. aeruginosa internalization [8]. The stimulation of internalization by exogenous Gg4 could be mediated via GM1 modulation of receptor kinase activity [55, 69] within lipid rafts required for internalization [46].
Serine Protease Activity
The protective effect of TLCK against binding may relate to early studies showing trypsin increased P. aeruginosa buccal cell attachment [70]. However, AEBSF was ineffective, although this serine protease inhibitor prevents protozoan host cell invasion [71]. The protective effect of TLCK may thus be due to the additional inhibition of plasmin.
The type 1 plasma membrane protein, gp140 and its plasmin cleavage product, p80, are the major epithelial substrates for src family kinases [72] and are expressed in IB3-1 and S9 cells(in progress). Proteolytic cleavage of gp140 and src mediated tyrosine phosphorylation correlate with cell monolayer [72], and such wounding predisposes to P. aeruginosa infection [73] and internalization [74]. TLCK prevents gp140 cleavage and suramin increases gp140/p80 phosphorylation [72]. The molecular basis of P. aeruginosa host cell binding remains unclear [18] but gp140 provides an attractive candidate. Gp140 ligation induces clustering within lipid rafts [75] necessary for GM1 involved, src-mediated phosphorylation and signaling to alter cell adhesion GM1 binding by exogenous Gg4 could modulate this pathway.
The TLCK protective effect could also result from inhibitory action on NFκB. Our demonstration that the NFκB inhibitor PPM-18, prevented internalization and that TLCK prevented target cell binding as well as internalization, suggest both these activities could be involved. NFκB activation is observed within 0.25-1hr of P. aeruginosa exposure [76], which would be in the preincubation time-frame we used for TLCK and PPM-18, but longer could be required for the phenotypic response.
A model of P. aeruginosa host cell binding and internalization, consistent with our findings would include: 1) exogenous GSL modulation of GSL-dependent src family kinase activity within lipid rafts, 2) a serine protease sensitive protein (gp140?) required for P. aeruginosa host-cell binding and 3) kinase-mediated tyrosine phosphorylation cascade to activate NFκB dependent P. aeruginosa host-cell invasion.
P. aeruginosa internalization has been proposed as an important feature of pathogenesis since this induces target cell apoptosis to facilitate clearing of the organism, which is defective in the absence of functional CFTR [9]. Without clearance, epithelial cell synthesis of inflammatory mediators is upregulated to exacerbate pathology. We suggest GSL mediated manipulation of internalization may influence the pathology of P. aeruginosa infection.

