All published articles of this journal are available on ScienceDirect.

Microbial Diversity of Acidic Hot Spring (Kawah Hujan B) in Geothermal Field of Kamojang Area, West Java-Indonesia
Abstract
Microbial communities in an acidic hot spring, namely Kawah Hujan B, at Kamojang geothermal field, West Java-Indonesia was examined using culture dependent and culture independent strategies. Chemical analysis of the hot spring water showed a characteristic of acidic-sulfate geothermal activity that contained high sulfate concentrations and low pH values (pH 1.8 to 1.9). Microbial community present in the spring was characterized by 16S rRNA gene combined with denaturing gradient gel electrophoresis (DGGE) analysis. The majority of the sequences recovered from culture-independent method were closely related to Crenarchaeota and Proteobacteria phyla. However, detail comparison among the member of Crenarchaeota showing some sequences variation compared to that the published data especially on the hypervariable and variable regions. In addition, the sequences did not belong to certain genus. Meanwhile, the 16S Rdna sequences from culture-dependent samples revealed mostly close to Firmicute and gamma Proteobacteria.
INTRODUCTION
Molecular studies of geothermal ecosystems have provided many illuminating discoveries of microbial diversity. Culturing methods and molecular studies have uncovered a remarkable diversity of both bacteria and archaea in hot springs worldwide. Microbes that are metabolically active in extremely thermoacidic environments have attracted for their unique ecology and physiology as well as sources of heat and acid stable biocatalysts [1]. Solfataric fields are the most important biotopes thermoacidophilic microbes. Most of thermoacidophilic microbes that usually lived in solfataric fields belong to the archaea including the genera Acidianus, Desulfurolobus, Metallosphaera, Stygiolobus, Sulfolobus, Sulfurisphaera, Sulfurococcus, Thermoplasma, and Picrophilus [1]. Microbes belong to bacteria were also found including Acidithiobacillus, Acidimicrobium, Sulfobacillus and Hydrogenobaculum genera [2, 3]. These chemolithotrophic acidophiles often are the predominant primary producers and may also contribute to iron and sulfur cycling via oxidization of reduced inorganic sulfur compounds [2-4].
The presence of diverse acidophilic populations in both natural and man-made acidic environments has been demonstrated by cultivation-dependent and -independent approaches [5-8]. However, the studies of microbial community from Indonesian geothermal fields are very limited [9, 10]. Indonesia is a country with a number of volcanoes and a lot of geothermal area. There are at least 120 volcanic centers that are spread over volcanic belts of 7000 km along the Indonesian islands [11]. Kamojang is one of these geothermal fields that located in West Java, Indonesia, at an altitude of 1500 m. The Kamojang geothermal field is the first operational geothermal field for electricity power in Indonesia [12].
The Kamojang geothermal field is vapor-dominated but the hydrothermal minerals show that the rock-altering fluid were dominantly liquid. The primary minerals present in the Kamojang subsurface rock samples are mainly feldspar (andesine-labradorite), pyroxene (hypersthene and augite), and olivine (forsterite). In general, the primary minerals in the andesite lavas are less altered than those in the pyroclastic rocks. There are two distinctive hydrothermal mineral assemblages at Kamojang, namely the acid and the neutral assemblages, which occur in shallower and deeper levels, respectively. The acid assemblage occupies the shallower level of the system (from near surface down to 100-300 m), and is characterized by the presence of kaolin with or without smectite, alunite, quartz, cristobalite, and pyrite. The deeper, neutral assemblages, comprises quartz, adularia, albite, epidote, titanite, wairakite, laumontite, calcite, siderite, titanohematite, pyrite, anhydrite, smectite, chlorite, illite, and interlayer clays [13].
The surface manifestations in the Kamojang area consist of hot pools, fumaroles, mud pots and hot springs lying in the so called Kawah Kamojang thermal area. Most of the hot surface water contains high concentration of sulfate (1000-2000 ppm) but low concentrations of chloride (< 5 ppm) [14]. The isotopic evidence suggests that the water was local meteoric water which has been heated by steam containing hydrogen sulfide, which oxidizes to sulfuric acid to give water of a low pH and high sulfate concentration [13].
Here we report the microbial community analysis of the acidic hot spring, namely Kawah Hujan B at Kamojang Geothermal Area, West Java, Indonesia. The analysis was based on culture-independent and culture-dependent strategies to get a first insight into the microbial communities in this acidic ecosystem. PCR amplification and DGGE separation of rRNA gene fragments were used to profile the microbial communities.
MATERIALS AND METHODS
Site and Sample Collection
The Kamojang geothermal field is located in West Java Province, Indonesia, about 35 km south of Bandung. Kawah Hujan B (E 107°48’14.38”, N -7°8’21.7”, the altitude 1690 m) is one of the hot springs in Kamojang Geothermal field. The hot spring is an acidic-sulfate mud pot. Water samples were collected in June 2006. For assessing microbial diversity by culture-independent method, water sample was filtered through a 0.22-µm-pore-size cellulose membrane filter (Sartorius, Germany) within 4 h after sampling. The cells on membrane were re-suspended in 25 ml of STE buffer (10 mM Tris-HCl [pH 8.0], 0.1 M NaCl, 1 mM EDTA) and precipitated by centrifugation. Pellet containing microbial communities were stored at -20°C until DNA extracted. Cultivation process was carried out by incubating spring water at 70°C after added by nutrients. Two nutrient composition were used as enrichment media, namely P (0.1% (w/v) peptone), and T (0.25% (w/v) tryptone; 0.25% (w/v) NaCl; 0.125% (w/v) yeast extract) media.
Geophysico-Chemical Analysis
pH and temperature were measured in June 2006. Measurement of cations concentration was performed using Atomic Absorption Spectroscopy/AAS (GBC Avanta Ver. 2.02) method and anions concentration was determined by titration, turbidimetry, spectrophotometry methods.
Bead Beating-Based DNA Extraction
The pellet containing microbial cells were mixed with DNA extraction buffer (100 mM Tris-HCl [pH 8.0], 100 mM sodium EDTA [pH 8.0], 100 mM sodium phosphate [pH 8.0], 1.5 M NaCl), glass beads and proteinase K (10 mg/ml) in microcentrifuge tubes by vortexing (Genie, G 560E, USA) at medium vigorous (half of maximum speed) for 15 min at room temperature. After mixing, 20% SDS was added, and the samples were incubated at 65°C for 2 h with gentle end-over-end inversions every 15 to 20 min. Supernatants were mixed with an equal volume of chloroform isoamylalcohol (24:1, v/v). The aqueous phase was recovered by centrifugation and precipitated with 0.6 volume of isopropanol at room temperature for 1 h. The pellet of crude nucleic acids were obtained by centrifugation at 16.000 g for 20 min at room temperature, washed twice with cold 70% ethanol, and resuspended in sterile deionized water.
Lysozyme-Based DNA Extraction
The pellet cells was suspended in 10 mM Tris HCl buffer (pH 8.0) containing 8 mg/ml of lysozyme and incubated at 37°C for 1 h, the cells were lysed by adding lysis buffer containing 2% SDS, 0.8 mg/ml proteinase K and 200 mM EDTA pH 8.0. The lysis process was carried out by incubation at 50°C for 30 min. Ice cold potassium acetate and acetic acid glacial mixed solution were added and the denatured proteins were precipitated by centrifugation. Supernatants were mixed with an equal volume of chloroform isoamylalcohol (24:1, v/v). The aqueous phase was recovered by centrifugation and precipitated with 0.6 volume of isopropanol at room temperature for 1 h. The pellet of crude nucleic acids were obtained by centrifugation at 16.000 g for 20 min at room temperature, washed twice with cold 70% ethanol, and resuspended in sterile deionized water.
Amplification of 16S rRNA Gene Fragments
The amplification of partial 16S rRNA genes was carried out as described previously by Ferris [15]. A set of primers were used to amplify about 323-bp section of the 16S rRNA genes of members of the domain Bacteria, including the highly variable V7-V8 region [16]. One primer complements a region conserved among members of the domain Bacteria (Escherichia coli positions 1055 to 1070; primer sequence 5’ATGGCTGTCGTCAGCT-3’). The other primer is based on a universally conserved region (E. coli positions 1392 to 1406; 5’CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCACGGGCGGTGTGTAC-3’) and incorporates a 40-base GC clamp. The specificity of this primer is imparted by the underlined region. PCRs were performed by using cloned Taq DNA polymerase according to the instructions provided by the manufacturer (Promega). The temperature cycle for the PCR was 1 min of denaturizing at 94°C, 1 min of annealing, and 1 min of primer extension at 72°C. During an initial touchdown cycle, the annealing temperature was continually decreased from 53 to 43°C in intervals of 1°C per cycle; 20 additional annealing cycles were performed at 43°C. The final primer extension was for 10 min at 72°C.
DGGE and re-PCR of DGGE Bands
DGGE was performed by using D-code systems (Bio-Rad Laboratories) with a 1.5-mm gel. Approximately 100- to 500-ng portions of PCR products were used directly onto 8% (w/v) polyacrylamide gels with denaturing gradients from 30 to 40% (100% denaturant was 7 M urea and 40% [v/v] deionized formamide). Electrophoresis was performed with 0.5X TAE buffer (20 mM Tris-acetate, 0.5 mM EDTA) at 200 V and 60°C for 4 h. After electrophoresis, the gels were stained with silver staining method [5]. Each band in the DGGE gel was excised with a razor blade, added with TE buffer (10mM Tris-Cl, 1 mM EDTA), placed in boiling water for 5 minutes, and then incubated overnight at 37°C. The eluted DNA was amplified using the same primers as previous amplification but without GC clamp sequence. PCRs were performed at the following conditions: an initial denaturizing step at 94°C for 5 min, followed by 30 cycles of 1 min at 94°C, 1 min at 50°C, and 1 min at 72°C, and a final extension step of 10 min at 72°C. All of confirm DGGE bands were subjected to DNA sequencing. Sequencing was carried out in an ABI PrismR 3100 Genetic Analyzer (Applied Biosystems) by the Macrogen Sequencing Service (Korea).
Phylogenetic Analysis
The sequencing results were compared to 16S rRNA gene sequences from GenBank database at NCBI (National Centre of Biotechnological Information) through web site http://www.ncbi.nlm.nih.gov using BLAST program [17] for screening of sequence similarity. Sequences alignments were performed by ClustalW program [18]. Aligning sequences were visualized using GenDoc program. Phylogenetic reconstruction was accomplished with the phylogeny inference package (PHYLIP version 3.62). Evolutionary distances were calculated by F84 method with the DNADIST program [19]. Phylogenetic trees were constructed from distance matrices by the neighbor-joining method [20], which was implemented with the NEIGHBOR program. The node reproducibility for tree topology was estimated by bootstrap analysis, which included 1000 replicate data sets. The trees were visualized by Treev32 program. Sequences were checked for chimeras using the CHECK_CHIMERA program of the Ribosomal Database Project [21].
RESULTS
Geochemical Characteristic
Kawah Hujan B has temperature at 90-92° C and pH 1.8-1.9. Chemical analysis of the hot spring water showed a high concentration of sulfate and Fe total ions. Some other metal ions, such as Mn, Pb, and Cu were detected in low concentration, as well anion such as chloride and nitrate (Table 1).
Concentrations of Total Soluble Constituents of Kawah Hujan B
| Analyte | Conc. mg/L |
|---|---|
| Ca | 9.48 |
| Mg | 0.23 |
| Fe | 11.81 |
| Mn | 0.22 |
| Na | 9.46 |
| K | 1.37 |
| Pb | 0.02 |
| Cu | 0.46 |
| Cl- | 32.67 |
| SO42- | 2,740.85 |
| Nitrate (NO3-) | 2.11 |
| Nitrite (NO2-) | 0.03 |
| Flouride (F-) | 0.63 |
Chromosomal DNA and DGGE Profile
Two DNA extraction methods, lisozyme-based and bead beating-based methods were performed independently to culture-independent (filtration) sample, while chromosomal DNA from culture-dependent (cultivation) samples were performed only by bead beating-based method. PCR amplifications of 16S rRNA gene fragments were successfully carried out for both filtration and cultivation samples. All of amplification results produced a single band in agarose gel electrophoresis with the size of approximately 400 bps.
DGGE profile of 16S rRNA gene fragments performed by filtration samples obtained from different cell lysis methods showed a different pattern. The different DGGE profiles were also performed by cultivation samples. Most of intense bands were appeared at the upper and lower sides of the gel (Fig. 1). However, some thin bands at the middle side of the gel were appeared on the filtration sample by enzymatically lysed and culture sample grown in P medium. Bands at the upper side of the gel showed a similar pattern. Most of the DGGE bands were successfully re-amplified and sequenced. The sequences estimated as chimeric artifact were excluded from further analysis.
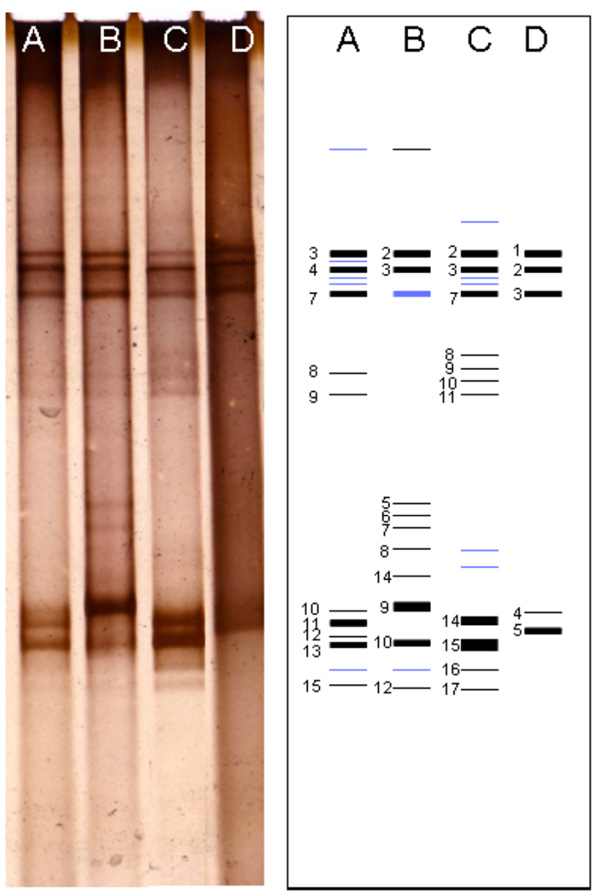
DGGE profiles of Kawah Hujan B samples. The individual bands that have been further analysis presented in number (1 - 17). Lane A-B, PCR product from filtration samples (bead-beating and lysozyme methods) respectively. Lane C-D, PCR product from cultivation samples (P and T media) respectively.
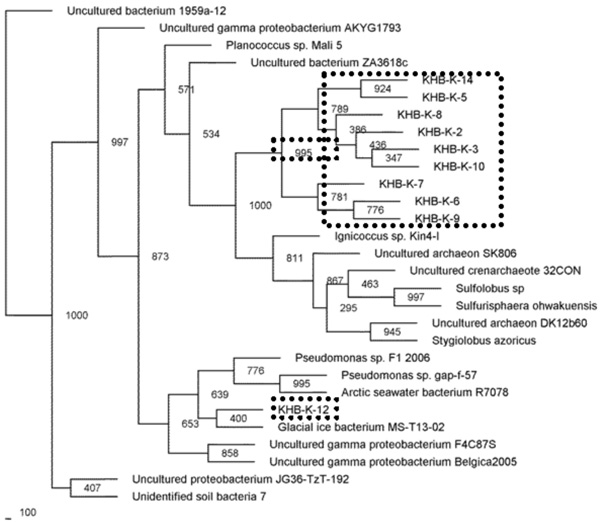
Phylogenetic relationships of the 16S rRNA gene fragment sequences obtained from the filtration samples using lysozyme cell lysis method and related sequences. Distance analysis was performed using the F84 correction, followed by phylogenetic tree construction using the neighbor-joining method of the PHYLIP software with 1,000 bootstrap replicates. (dashline), represented the samples.
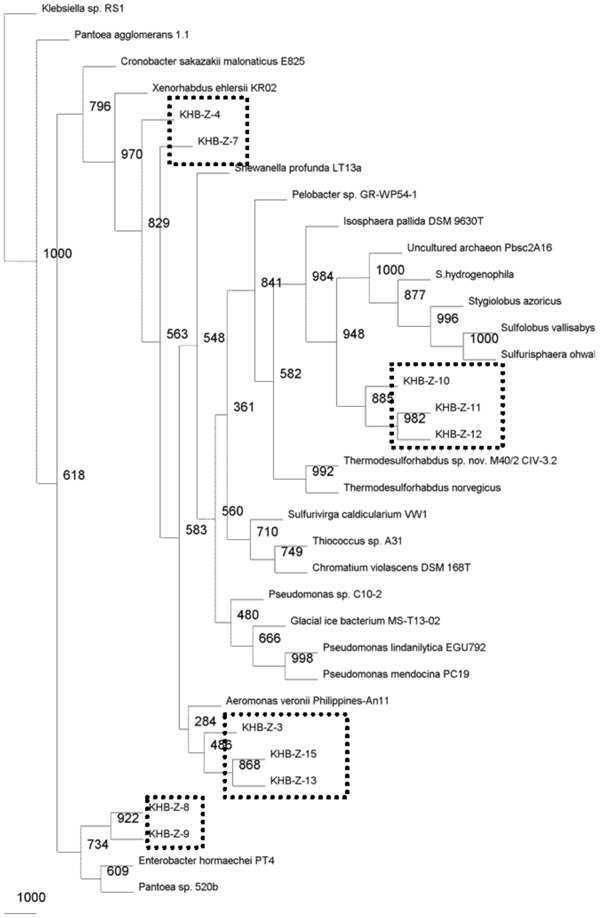
Phylogenetic relationships of the 16S rRNA gene fragment sequences obtained from the culture-independent samples using bead-beating cell lysis method and related sequences. Distance analysis was performed using the F84 correction, followed by phylogenetic tree construction using the neighbor-joining method of the PHYLIP software with 1,000 bootstrap replicates. (dashline), represented the samples.

Comparison of nucleotide sequences of DGGE band from culture-independent sample. A. Close to Crenarchaeota groups. B. Close to gamma Proteobacteria groups. (Grey block) represented hypervaribale region (V7 and V8); (.), represented same bases.
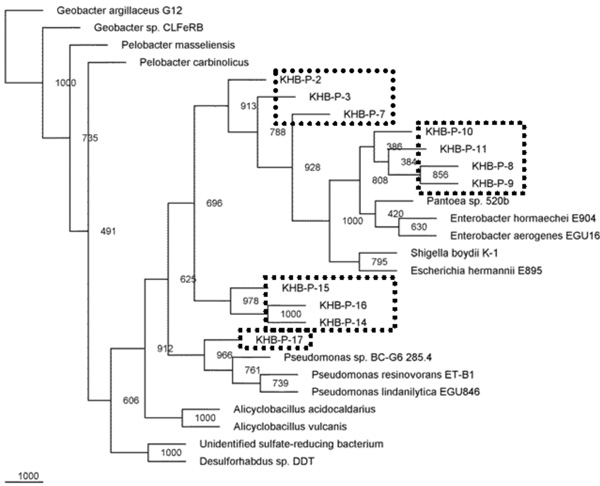
Phylogenetic relationships of the 16S rRNA gene fragment sequences obtained from the culture grown in P medium and related sequences. Distance analysis was performed using the F84 correction, followed by phylogenetic tree construction using the neighbor-joining method of the PHYLIP software with 1,000 bootstrap replicates. (dashline), represented the samples.
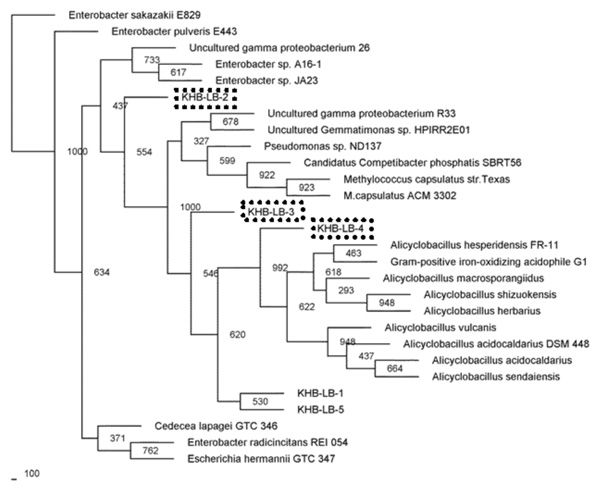
Phylogenetic relationships of the 16S rRNA gene fragment sequences obtained from the culture grown in T medium and related sequences. Distance analysis was performed using the F84 correction, followed by phylogenetic tree construction using the neighbor-joining method of the PHYLIP software with 1,000 bootstrap replicates. (dashline), represented the samples.

Comparison of nucleotide sequences of DGGE band which is close to Alicyclobacillus (A) and Enterobacter (B) sequences. (Grey block) represented for hypervaribale region; (.), represented same bases.
Phylogenetic and Diversity of Community
Microbial community present in acidic-sulfate containing hot spring was characterized by 16S rRNA gene fragment analysis. The majority of the sequences recovered from filtration sample obtained by enzymatic-based cell lyses method were affiliated with the Crenarchaeota phylum, and only one sequence was closed to Pseudomonas genus belonging to Proteobacteria phylum (Fig. 2). While the sequences recovered from filtration sample obtained by bead-beating method were performed slightly different lineage on phylogenetic tree (Fig. 3). Sequences affiliated with the Crenarchaeota were minority in filtrated sample obtained by bead-beating method. Most of sequences were close to gamma Proteobacteria group including Aeromonas, Xenorhabdus, and Enterobacter genera. In phylogenetic tree, the sequences closed to Crenarchaeota showed clustering in a distinct branch (Fig. 2).
Detail alignment of the sequences to closely related sequences from database showed that the sequences belonged to Crenarchaeota group showed many bases substitution compared to that the sequences available in the GenBank database (Fig. 4A). Sequences variations were observed throughout the region that lied in the variable region as well as conserved region. Variation sequences were also observed on the sequences that closed to gamma Proteobacteria group. However, most of the differences were laid in the V7 and V8 hypervariable region (Fig. 4B), particularly for the sequences closed to Aeromonas genus. Meanwhile the sequences closed to Pantoea/Enterobacter genus had relatively similar sequences compared to that the sequences available in the GenBank database (data not shown).
Microbial diversities from the cultivation samples were mostly different with the filtration sample. The sequences recovered from cultivation in P medium revealed as gamma Proteobacteria. Most of them were affiliated to Pantoea/Enterobacter genus, three sequences performed distinct cluster with other known microbes, and one sequences closed to Pseudomonas (Fig. 5). While, most of the sequences detected in T medium were closed to Alicyclobacillus, and only one sequence was affiliated with gamma proteobacterium (Fig. 6).
Detail alignment for the sequence close to Pseudomonas genus revealed slightly differences with other sequences from the GenBank database. Most of sequence variation lied in variable region and one nucleotide difference detected in conserved region. While detail alignment for the sequences obtained from T medium that closed to Alicyclobacillus showed sequence variation, particularly in V7 and V8 hypervariable region (Fig. 7).
DISCUSSION
The Kawah Hujan B of Kamojang geothermal filed is a stressfull, natural environment with an extremely low pH, high temperature and high load of sulfate ion. Investigation of microbial diversity using culture-dependent and culture-independent revealed that the microbes from culture-dependent were belong to Crenorchoeota and Proteobacteria phyla while from culture-dependent belong to Firmicute and gamma Proteobacteria.
The present of Crenorchaeota in Kawah Hujan B was predicted since these organisms frequently found in extreme environment [22, 23]. Some species of Crenorchaeota are thermoacidophile growing in environment with high sulfate concentration [22]. Crenorchaeota was aerob, facultative anearob, and anaerob, chemoorganotroph and lithotroph, and able to use sulphur as energy source in their metabolism [24, 25]. In this experiment, most of microbes detected by culture-independent strategy were archaea suggesting that the Kawah Hujan B was suitable habitat for these microbes. A few reports showed that archaea were predominant microbes on the environments with extreme temperature (>90°C) [26], and most of thermoacidophile archaea were belonging to Crenorchaeota phylum [27]. Furthermore, some hyperthermophilic archaea were known as reducing sulfur organisms [28]. Some genus of Crenorchaeota, such as Sulfolobus dan Acidianus were known as sulfur oxidicing microbes [29, 30]. High concentration sulfate ion in Kawah Hujan B indicated that there were sulphur oxidicing and reducing microbes [22]. However, homological analysis of 16S rRNA gene fragment of Crenorchaoeta from Kawah Hujan B showed that the sequences have many based substitution compared to that the GenBank sequences. In addition, the phylogenetic analysis showed that the organisms clustering in different branches. This suggested that the Crenorchaoeta from Kawah Hujan B might be distinct with other known organisms.
The microbes, obtained from enzymatic based-cell lyses were slightly different with physical lyses. Aeromonas and Pantoea/Enterobacter were only detected by physical based method while microbe belongs to Pseudomonas only found on sample lyses by enzymatic method, while, the archaea were detected by both methods. This again suggested that Kawah Hujan B is suitable for archaea niches. The fail to detect Aeromonas and Pantoea by enzymatic based lyses but successfully by physical treatment suggested that organism might form biofilms or spores in Kawah Hujan B. Formation of biofilm were one of microbial adaptation for dynamic condition of environment [26]. Furthermore, some microbes required to form biofilm for growth on the surface, especially for mud-associated growth mcrobes [31].
In this study some microbes found by culture-independent were not detected in cultivation samples. This is probably due to some predominant microbes in natural samples belong as unculturable organisms, while microbes were less dominat in nature could grow better in compatible media. The presence of Firmicute, such as Alicyclobacillus in Kawah Hujan B is not surprising since this microbe often found in geothermal region [32-35]. Alicyclobacillus was thermoacidophilic microbe, Gram-positive and endospore forming [32, 36, 37]. Alicyclobacillus was aerobic and some showed several phenotypic properties such as the ability to oxidize iron, elemental sulfur and sulfides [34, 36]. These conditions were provided by Kawah Hujan B.
CONCLUSIONS
Biodiversity of microbial diversity from an acidic hot spring at Kamojang Geothermal Field was performed based on culture-dependent and culture-independent strategies. The majority of microbes revealed from the culture-independent were close to Chrenorchaeota and Proteobacteria, however, detail analysis of the sequences closed to Chernorchaeota showed difference with other known sequence from the genbank. Meanwhile microbes identified by culture-dependent samples were closed to Firmicute and gamma Proteobacteria.
ACKNOWLEDGEMENTS
This research was funded by grants from ITB Research Program to Akhmaloka and Incentive of Fundamental Research from Ministry of Research and Technology, Republic of Indonesia to Fida Madayanti, and BPPS Scholarship to Heni Yohandini.

