All published articles of this journal are available on ScienceDirect.

Antibiofilm and Molecular Effects of Ceftriaxone-loaded PLGA Nanoparticles Against Clinical Haemophilus Influenza
Abstract
Introduction
Haemophilus influenzae biofilm-related infections are a significant issue of therapeutic challenge because these infections are resistant to antibiotics and associated with a high recurrence rate. Nanotechnology in drug delivery systems can help improve the antimicrobial efficacy through better penetration and sustained release in biofilms.
Methods
The development of a ceftriaxone-loaded poly (lactic-co-glycolic acid) (PLGA) nano-delivery system was done through the double-emulsion solvent evaporation procedure. Nanoparticles have been characterized in terms of size, morphology, and encapsulation efficiency. Minimum inhibitory concentration (MIC) testing and biofilm assays were used to measure the antimicrobial activity. Crystal violet staining was employed to measure biofilm biomass, whereas colony-forming unit (CFU) was used to measure bacterial viability. At 6 and 24 hours, RT-qPCR was done on adhesion/ biofilm-related genes (hia, hmw, hif, luxS) and resistance/ stress genes (blaTEM, blaROB-1, ftsI, acrB).
Results
PLGA-ceftriaxone formulation was found to have better antibiofilm activity than free ceftriaxone. A sharp decrease in biofilm biomass and viable cell counts was observed, and a relationship was found between biofilm inhibition and decreased bacterial viability. The RT-qPCR result showed the down-regulation of adhesion and quorum-sensing genes, as well as the overall decrease in the resistance and stress-related gene expression, especially at 24 hours.
Discussion
It can be inferred that the enhancement in the performance of the nano-formulation can be explained by the sustained drug release, increased penetration into the extracellular polymeric matrix, and extended exposure to localized antibiotics.
Conclusion
Ceftriaxone-loaded PLGA nanoparticles should be further investigated in in vivo and translational studies as they may be an effective in vitro antibiofilm delivery method against clinical H. influenzae isolates.
1. INTRODUCTION
According to recent global assessments, bacterial resistance is already linked to a significant burden of mortality and is expected to increase pressure on health systems in the coming decades, making antimicrobial resistance one of the greatest threats to modern medicine [1]. From a clinical and public health standpoint, the introduction of difficult-to-treat respiratory bacterial pathogens is especially significant since lower respiratory infections continue to be a major source of morbidity and death globally [2].
Haemophilus influenza (H. influenza), particularly non-typeable strains, remains clinically significant as a respiratory pathogen, linked to mucosal illnesses, recurrent otitis media, chronic obstructive pulmonary disease flare-ups, and persistent airway infections. Its capacity to form biofilm communities, which improve resistance to host defenses and antimicrobial treatment, is a key factor in its persistence [3]. H. influenzae biofilms are associated with persistent colonization and decreased antibiotic susceptibility, according to earlier research, which makes this organism a good pathogen-specific model for assessing enhanced local drug-delivery techniques. Furthermore, decreased β-lactam susceptibility may also result from changes in penicillin-binding protein 3 encoded by ftsI, including the well-known BLNAR-associated pathway [4]. This means that resistance in H. influenzae is not restricted to traditional β-lactamase synthesis.
Antibiotic delivery using nanoparticles has been extensively studied as a method to increase penetration into biofilm-associated illnesses, maintain drug release, and improve antimicrobial exposure [5]. Poly(lactic-co-glycolic acid) (PLGA) is one of the most researched biodegradable polymers for drug delivery applications, due to its biocompatibility, controlled drug release behavior, and ability to contain antibacterial drugs [6]. By enhancing local delivery and extending drug availability, PLGA-based nano-platforms have demonstrated promise in anti-biofilm applications. Nevertheless, the majority of reports discuss the PLGA-based nano-platforms concept in general, while there are still fewer pathogen-focused studies that combine microbiological, biofilm, and H. influenzae isolates. Therefore, determining whether a ceftriaxone-loaded PLGA system may provide quantifiable benefits against a clinically relevant H. influenzae biofilm model is more valuable than claiming a generally novel nano-antibiotic concept [7, 8].
In light of this reasoning, the current investigation assessed the antibacterial and antibiofilm properties of PLGA nanoparticles loaded with ceftriaxone against clinical isolates of H. influenzae. In order to investigate whether the nano-formulation enhanced antimicrobial performance as well as whether it changed bacterial responses related to persistence and treatment failure, the work combined minimum inhibitory concentration testing, complementary biofilm assessment, and RT-qPCR analysis of specific virulence- and resistance-related genes. This research was conceived as a targeted proof-of-concept examination of clinically relevant respiratory biofilm pathogens.
The novelty of this study does not lie in presenting polylactic-co-glycolic acid (PLGA)-based antibiotic delivery systems as a novel anti-biofilm platform, although PLGAs have been extensively studied. Rather, this study, using therapeutically important H. influenzae isolates provide valuable practical evidence of the viability of this technique with a focus on specific pathogens. This is achieved through a combination of physicochemical characterization, time-dependent RT-qPCR analysis of virulence and resistance-associated genes, dual biofilm readings (crystal violet staining and colony-forming unit count), and antibiotic efficacy comparison with the active drug. The study's contribution in this regard is that it demonstrates that ceftrexone-loaded PLGA nanoparticles are associated with a broader downregulation of adhesion, quorum sensing, and resistance/stress genes, as well as enhanced antibiotic efficacy compared to free ceftrexone. This develops our knowledge beyond simply improving the delivery process to include a more comprehensive understanding of the antimicrobial response mechanism.
2. LITERATURE REVIEW
Haemophilus influenzae atypical (NTHi) is a respiratory pathogen that may build biofilms and transition from colonization of mucous membranes to chronic infection. This biofilm confers greater survival, reduces treatment efficacy, and contributes to disease persistence [9]. The literature also indicates that the dynamics of host-NTHi interaction-including mucosal adhesion, colonization, and immune evasion-are fundamental to the transition from commensal to pathogenic, with the biofilm playing a prominent role as a protective and persistent environment for infection [10]. More recent reviews support a detailed understanding of the biofilm formation mechanisms in H. influenzae and their relationship to clinical outcomes, including difficulty in eradication and recurrence, making the biofilm a logical target for therapeutic interventions [5].
At the level of virulence factors associated with adhesion and biofilm, adhesion systems (such as hia and the HMW system) are important determinants of NTHi's attachment to epithelial cells and the formation of early biofilm structure. Studies show that disruption/reduction of adhesion components is reflected in colonization capacity and microbial community cohesion [5, 10]. Regulatory signals associated with cell communication are also linked to biofilm maturation and the consistency of microbial community behavior, making their gene expression tracking useful as an indicator of biofilm “maturity” and response to treatment [5]. Furthermore, immunological targeting of biofilm matrix components in NTHi may alter the biofilm-associated phenotype and increase the cells' susceptibility to transition to a more fragile state after biofilm release, supporting the immunological importance of matrix components as therapeutic/supportive targets [11].
Regarding antibiotic resistance, recent literature indicates that NTHi resistance to beta-lactams is influenced by two main axes: β-lactamase production (e.g., blaTEM and blaROB-1) and PBP3/ftsI alterations, which are associated with reduced sensitivity to beta-lactams even in the absence of β-lactamase in some phenotypes [12]. Furthermore, extensive epidemiological data show that ampicillin/amoxicillin-clavulanate resistance may be linked to β-lactamase production across multiple years and geographically broad ranges, explaining the need for more reliable beta-lactam options in certain clinical settings [13]. Molecular surveillance studies support the association of resistance patterns with local genetic/epidemiological characteristics, making the inclusion of genes such as blaTEM, blaROB-1, ftsI, and ejection pump markers (e.g., acrB) a logical inclusion within a “molecular fingerprint” of the response under drug stress [14]. Recent regional reports also describe the clinical/laboratory characterisation of ceftriaxone-resistant or relatively low-susceptibility isolates within paediatric populations, emphasising the importance of monitoring emerging susceptibility patterns when selecting an antibiotic model [15]. Typical laboratory point of view. For routine susceptibility testing (broth microdilution, etc.), sources suggest following established procedures for analysis and comparison of MICs between treatments [16].
PLGA polymers are among the most widely used drug delivery systems in the field of nanosolutions, thanks to their biocompatibility and ability to control drug release and degradation. According to specialized studies, PLGA antimicrobial platforms may enhance efficacy by improving local bioavailability and prolonging the effective exposure period, particularly in chronic or biofilm-associated diseases [17]. Given that H. influenzae infections are often respiratory in origin, studies suggest that PLGA systems (particularly PLGA-PEG) in pulmonary administration may provide notable benefits in respiratory tract infections [18]. The advantage of some nanostructured formulations over free drugs in mature biofilm models can be explained by the fact that the properties of the particles (size, dispersion, and surface charge) directly affect their interaction with the biofilm matrix and their penetration capabilities [17, 19]. The presence of an “EPS penetration” mechanism as a crucial component in enhancing biofilm control efficacy when optimizing particle design is supported by a study on PLGA particles [20].
The water/oil/water dual emulsion method is widely used to encapsulate hydrophilic drugs in lactic-co-glycolic acid (PLGA) polymers, and has been systematically supported in recent work focusing on encapsulating antimicrobials/compounds within nanocarriers via dual emulsion to achieve proper containment and controlled release [21]. Extensive polymer reviews also summarize that PLGA-NPs with antimicrobial properties have demonstrated applicability in multiple contexts, emphasizing the relationship between design (formulation/stabilization/emulsion) and therapeutic outcome [22].
Regarding biofilm evaluation, Crystal Violet (CV) measurement for biomass estimation is widely used, but combining CV and CFU provides a dual “structural + bioss” reading that reduces the risk of interpreting a decrease/increase in biomass without corresponding bioss, or vice versa [23]. Recent biofilm measurement literature also supports the importance of comparable operational definitions, such as MBEC, when studying eradication, and emphasizes that the choice of measurement instrument/design affects the comparability of results between studies [24]. Consequently, integrating CV/CFU indices with sensitivity criteria (MIC) within a free-drug and nano-loaded drug comparison design represents a methodologically sound approach for investigating the effects of “exposure pattern” and drug release on biofilm [17, 18]
At the molecular level, real-time quantitative polymerase chain reaction (RT-qPCR) is a common tool for monitoring the “regulatory fingerprint” of bacterial response under therapeutic stress. MIQE 2.0 recommendations emphasized the need to document No-RT/NTC controls, verify the stability of reference genes, and report amplification efficiency to avoid misleading conclusions [25, 26]. Furthermore, the use of early and late time points in gene expression studies is employed to capture rapid regulatory responses that might not be apparent if the measurement were limited to a single late point-a methodological rationale consistent with time-course designs in molecular studies [24]. Lastly, the literature highlights that in order to establish causality, changes in resistance genes (such as blaTEM/blaROB-1, ftsI, and efflux pumps) must be correlated with prototyping assays (β-lactamase) or characterizing mutations (PBP3/ftsI) rather than depending only on gene expression [12, 16].
3. MATERIALS AND METHODS
3.1. Study Design
A comparative pilot study was conducted to evaluate a biodegradable polymer nano-delivery system (PLGA or PLGA-PEG) containing an antibiotic aimed at H. influenzae. The study included a direct comparison with a free drug and a negative control, with empty PLGA particles serving as an additional control to separate the polymer from the medication [22].
Ceftriaxone was chosen as the model antibiotic for two methodological reasons: (1) the increasing resistance of H. influenzae to ampicillin due to β-lactamase and PBP3/ftsI modifications, necessitating more reliable alternatives; and (2) the ability of ceftriaxone (as a hydrophilic compound) to be effectively integrated into PLGA via W/O/W with controlled release [12, 13, 27].
During the research period, 12 clinical isolates of Haemophilus influenzae were obtained from Ramadi General Hospital; these isolates were laboratory-confirmed before inclusion. Initially, all isolates were examined for their capacity to form biofilms and their sensitivity to antibiotics. Instead of employing a single strain, many clinical isolates were used for the phenotypic tests, which included MIC determination, time-kill analysis, biofilm inhibition, mature biofilm eradication, and CFU-based viability evaluation.
Three typical multidrug-resistant isolates were chosen for further RT-qPCR study based on the initial resistance profile. These isolates were chosen because they were thought to be indicative of the resistant phenotype under investigation in this research and decreased sensitivity to β-lactam therapy. Both β-lactamase-associated and non-β-lactamase-mediated markers were used to characterize resistance. While ftsI was added as a marker linked to altered penicillin-binding protein 3 and BLNAR-related decreased β-lactam resistance, blaTEM and blaROB-1 were included as β-lactamase-associated determinants. AcrB was also included as a marker linked to efflux and stress response. In order to offer an exploratory molecular profile of the response to ceftriaxone-loaded PLGA nanoparticles, RT-qPCR analysis was carried out on a typical multidrug-resistant subgroup in the current investigation, which included numerous clinical isolates for phenotypic assessment.
Following initial phenotypic screening of all 12 clinical isolates, the three isolates were chosen as a representative resistant subset for exploratory molecular profiling; this strategy was meant to provide a targeted proof-of-concept molecular readout while remaining practical within the parameters of the pilot study.
The MIC test was used with a modern, standardized method (broth microdilution) to obtain the baseline for how sensitive the free drug is to other drugs. This way, we could compare the amounts of the free drug and the nanomedicine by using the active ingredient equivalent [16]. Table 1 shows the materials used in the current study.
| Category | Material/Reagent | Brief Specification | Company | Applications |
|---|---|---|---|---|
| Nanopolymer | PLGA (Resomer® RG 503 H) | PLGA 50:50, acid-terminated | Sigma-Aldrich | Preparation of PLGA-NPs |
| Nanopolymer (alternative) | PLGA-PEG (PEG-b-PLGA) | mPEG-PLGA (suitable for improved stability/permeability) | Sigma-Aldrich | Preparation of PLGA-PEG-NPs |
| Emulsifier | Poly(vinyl alcohol) (PVA) | 87–89% hydrolyzed | Sigma-Aldrich | W/O/W Emulsification Stabilizer |
| Antibiotic | Ceftriaxone disodium salt hemihydrate | Disodium salt | Sigma-Aldrich | Free Drug + Loading into PLGA |
| Buffering Solution | PBS, pH 7.4 (1X) | No Ca/Mg | Gibco (Thermo Fisher) | Washing/Suspension/Solution Preparation |
| Stabilizer/Protector (optional) | Trehalose dehydrate | ≥99% (depending on availability) | Sigma-Aldrich | Particle Stabilization during Drying/Storage |
| RNA Precipitation/Purification | Ethyl alcohol, 200 proof (MB grade) | Suitable for RNA/DNA extraction | Sigma-Aldrich | Precipitation/Washing Step in RNA Protocols |
| Biofilm Measurement | Crystal violet solution | Crystal violet solution (HT901) | Sigma-Aldrich | Biofilm Biomass Measurement |
| H. influenzae Growth Medium/Factors | Hemin | Factor X | Sigma-Aldrich | Growth Stabilization (Factor X) |
| H. influenzae Growth Medium/Factors | β-NAD (Nicotinamide adenine dinucleotide) | Factor V | Sigma-Aldrich | Growth Stabilization (Factor V) |
| RNA Extraction Kit | RNeasy Mini Kit (50) | Silica columns | QIAGEN | High-Quality RNA Extraction |
| Genomic DNA Removal | RNase-Free DNase Set (50) | DNase I + RDD buffer | QIAGEN | Prevention of DNA Contamination before RT-qPCR |
| cDNA Synthesis | QuantiTect Reverse Transcription Kit | Includes gDNA removal | QIAGEN | RNA to cDNA Conversion |
| qPCR Master Mix (SYBR) | QuantiNova SYBR Green PCR Kit | 2× Master Mix | QIAGEN | RT-qPCR (SYBR) |
| qPCR Master Mix (alternative) | PowerUp™ SYBR™ Green Master Mix | 2× Master Mix | Applied Biosystems (Thermo Fisher) | SYBR Alternative to RT-qPCR |
| Nuclease-Free Water | Nuclease-Free Water | Not DEPC-treated | Ambion/Invitrogen (Thermo Fisher) | Preparation of RT/qPCR Mixtures |
| qPCR Plates | MicroAmp™ Optical 96-Well Plate | 96-well | Applied Biosystems | RT-qPCR Reactions |
| qPCR Optical Adhesive | MicroAmp™ Optical Adhesive Film | 100 covers | Applied Biosystems | QPCR Plate Sealing and Evaporation Prevention |
| 96-well Plates (biofilm/MIC) | 96-well plate flat-bottom | Polystyrene | Corning | MIC/Crystal Violet Biofilm |
| Dialysis Membrane/Tube (optional for release) | Dialysis tubing 12–14 kD MWCO | For the release study | Spectra/Poor (Repligen) | Drug Release Testing from Particles |
Note: PLGA = poly (lactic-co-glycolic acid); PLGA-PEG = poly (lactic-co-glycolic acid)-polyethylene glycol; PVA = poly (vinyl alcohol); PBS = phosphate-buffered saline; RNA = ribonucleic acid; DNA = deoxyribonucleic acid; DNase = deoxyribonuclease; cDNA = complementary DNA; SYBR = SYBR Green fluorescent dye; RT-qPCR = real-time quantitative polymerase chain reaction; MIC = minimum inhibitory concentration. Materials are listed with their main analytical or experimental use in the study.
3.2. Preparation and Characterization of Ceftriaxone-loaded Nanoparticles
Because PLGA or PLGA-PEG nanoparticles are a popular Nano delivery technology with biocompatibility, adjustable breakdown, and the possibility for continuous antibiotic release, they were used. In comparison to free-form medications, this is anticipated to boost local bioavailability and enhance antibacterial activity [17, 18].
3.2.1. Preparation Method (Double Emulsion W/O/W) of Ceftriaxone
Because ceftriaxone is hydrophilic, an emulsion was used. This technique is advised for managing volume and release while optimizing the aqueous drug concentration inside PLGA [21, 22].
In actuality, PLGA (or PLGA-PEG) was dissolved in an appropriate organic solvent to create the O phase, and ceftriaxone was dissolved in the internal aqueous phase W1. After that, a W1/O emulsion was homogenized and sonicated under uniform circumstances for every batch. W1/O/W2 was created by pouring this emulsion into an external aqueous phase W2 that included an emulsifier (like PVA). After that, the solvent was eliminated by swirling and evaporating the particles until they formed. Following centrifugation, the particles were collected and cleaned to get rid of any leftover medication and emulsion residue [22].
3.2.2. Physicochemical Characterization
The physicochemical characterization of the prepared nanoparticles included particle size, polydispersity index (PDI), zeta potential, and morphological examination. Particle size distribution, PDI, and zeta potential were measured using dynamic light scattering techniques to assess formulation uniformity and colloidal behavior. In addition, nanoparticle morphology was examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) to confirm particle shape, surface characteristics, and aggregation pattern. To evaluate formulation reproducibility, the nanoparticles were prepared in three independent batches, and the measurements were recorded as mean ± SD. Short-term stability was assessed by monitoring particle size, PDI, and zeta potential during storage under refrigerated conditions over a defined period.
Containment efficiency (EE%) and drug load (DL%) were calculated by measuring the free drug in the filtrate or by dissolving the particles and measuring the content (UV-Vis/HPLC, as available), since EE/DL is the basis for determining the “equivalent dose” between the nanomedicine and the free drug in efficacy and RT-qPCR assays [22].
3.2.3. In Vitro Release Study
Using a suitable release medium and constant stirring, the in vitro release profile of ceftriaxone from PLGA nanoparticles was assessed at 37°C. Samples were taken at pre-arranged intervals, and their total ceftriaxone release was examined. The Higuchi and Korsmeyer–Peppas kinetic models were fitted to the experimental release data to better describe the release process. While the Korsmeyer-Peppas model was employed to determine whether the release mechanism followed Fickian diffusion, anomalous transport, or polymer relaxation/erosion-associated release behavior, the Higuchi model was used to assess diffusion-controlled release from the polymeric matrix [17].
3.3. Evaluation of Antibacterial Activity and Biofilm Effect
After standardizing the inoculum density, the MIC of free drug and PLGA-Ceftriaxone (with empty particles as a control) was performed on an API-equivalent basis, with any differences documented as reflecting improved delivery/release rather than a difference in the amount of active ingredient [16, 22].
A time-kill experiment was also performed at equivalent concentrations (e.g., 1× and 2× MIC) to compare the time-kill dynamics between free drug and nanomedicine, since the sustained release effect is typically more clearly observed over time than at a single point [22].
Because biofilm formation is a key factor in the persistence of H. influenzae infection and the decline in antibiotic response, the following were measured:
(a) Inhibition of biofilm formation by exposing bacteria to treatments during the formation stage, and (b) Eradication of mature biofilms after pre-formation, with biomass estimation by crystal violet and/or bacterial count (CFU) of the deconstructed biofilm, since combining CV and CFU yields a “structure + biotic” reading [5, 24].
To define MBEC/MBEC90 comparably, standard MBEC measurement systems/concepts described in recent literature for high-throughput biofilm instruments can be utilized [24].
3.4. RT-qPCR Molecular Fingerprinting of Virulence and Resistance Genes and their Correlation with Release Pattern
RT-qPCR was carried out in compliance with MIQE-based reporting guidelines. Bacterial samples were taken at 6 and 24 hours after exposure to either free ceftriaxone or ceftriaxone-loaded PLGA nanoparticles at equal active-drug concentrations to record early and late transcriptional responses. To reduce genomic DNA contamination, total RNA was extracted using a bacterial RNA extraction procedure with an on-column DNase treatment. Before cDNA synthesis, the amount and quality of RNA were evaluated [25, 26].
Standardized RNA input quantities were used to create cDNA, and No-RT controls were added to ensure that genomic DNA contamination was not present. In order to identify potential reagent contamination or nonspecific amplification, no-template controls (NTCs) were included in every qPCR run. Target genes included those linked to biofilm and virulence (hia, hmw1A, hmw2A, hifA/hifBC, iga, hap, luxS, and ompP2) and resistance/stress (blaTEM, blaROB-1, ftsI, and acrB). Before normalization, the expression stability of candidate reference genes (rpoD, gyrA, and recA) was evaluated across treatment groups and time periods [12, 14, 27].
Melt-curve analysis was utilized to verify primer specificity, and gene-expression analysis was limited to primer pairings that produced a single distinct melt peak. Standard curves made with serially diluted cDNA were used to determine amplification efficiency, and primer pairs with acceptable efficiency and linearity were kept. The 2^-ΔΔCt technique was used to determine relative gene expression. For every experimental group, three independent biological replicates were used for RT-qPCR analysis; each biological replicate was examined in a technical triplicate [5, 12, 24, 28].
3.5. Statistical Analysis
To confirm the appropriateness of parametric analysis, the data were checked for near normality and homogeneity of variance before running the unpaired Student's t-test or one-way ANOVA. Only after ensuring that these presumptions were adequately satisfied were parametric tests used for statistical comparisons.
All data were analyzed using GraphPad Prism version 10.0. Physical and chemical characterization experiments, including particle size, dispersion index, zeta potential, encapsulation efficiency, and in vitro release testing, were performed using three independently prepared synthetic batches (n = 3). Microbiological assays, including minimum inhibitory concentration (MIC), minimum growth inhibitory concentration (MBIC), minimum growth inhibitory concentration (MBEC), time-kill test, coefficient of variation (CV), and bacterial colony count (CFU) analysis, were performed using three independent biological samples (n = 3). Real-time quantitative polymerase chain reaction (RT-qPCR) experiments were performed using three independent biological samples for each experimental group, and each biological sample was analyzed in triplicate. Results are presented as mean ± standard deviation (SD).
For comparison between the two groups, an unpaired Student’s t-test was used. To compare three or more groups, one-way analysis of variance (ANOVA) was performed, followed by Tukey's post-hoc test for multiple comparisons. In RT-qPCR analysis, relative expression values were calculated using the 2^-ΔΔCt method after calibration with the approved reference gene, and a statistical test was performed on the calibrated biological transcript data. A p-value<0.05 was considered statistically significant [25, 26].
3.6. Research Ethics and Ethics Committee Approval
This study received approval from the Ethics Committee of [University of Anbar /College of Science] under official letter number [154] dated [3/11/2025], and the necessary approvals were obtained from the relevant clinical authorities. When dealing with human samples, informed consent was obtained (where applicable), with samples coded, any personal identifiers removed, and data access restricted to authorized persons only.
4. RESULTS AND DISCUSSION
4.1. Nanoparticle Characterization, Containment Efficiency, and Drug Load
Ceftriaxone-loaded PLGA particles with the right size and dispersion properties for use against biofilms were created using the W/O/W double emulsification technique. In line with the study's goal of enhancing delivery in comparison to free medication, the anticipated negative surface charge of the PLGA polymer adds to stability and contact with the extracellular matrix, as shown in Table 2.
| Parameter | PLGA-Ceftriaxone | Blank PLGA |
|---|---|---|
| Mean size (nm) | 172.4 ± 9.8 | 154.6 ± 8.5 |
| PDI | 0.18 ± 0.03 | 0.16 ± 0.02 |
| Zeta potential (mV) | −21.7 ± 2.4 | −23.1 ± 2.1 |
| Encapsulation efficiency (%) | 63.2 ± 4.7 | None |
| Drug loading (%) | 7.9 ± 0.7 | None |
Note: Data are presented as mean ± SD of three independently prepared batches (n = 3). PLGA-Ceftriaxone = ceftriaxone-loaded PLGA nanoparticles; PLGA = blank PLGA nanoparticles; PDI = polydispersity index; EE% = encapsulation efficiency; DL% = drug loading. EE% and DL% were calculated only for the drug-loaded formulation.
Physical characterization results showed that the ceftriaxone-loaded nanoparticles (PLGA-Ceftriaxone) were larger than the blank nanoparticles (PLGA), with an average size of 172.4 ± 9.8 nm versus 154.6 ± 8.5 nm. This increase in size indicates successful drug loading within or onto the PLGA matrix, as an increase in hydrodynamic diameter after loading is a common indicator of active ingredient incorporation within the Nano system.
As for the PDI, values were low in both systems (0.18 ± 0.03 for the loaded and 0.16 ± 0.02 for the blank), indicating a relatively homogeneous size distribution and low dispersion. Furthermore, the difference between the two groups appears to be minimal, suggesting that ceftriaxone loading did not negatively affect particle homogeneity.
Regarding the zeta potential, the values were negative in both preparations, reaching −21.7 ± 2.4 mV in PLGA-Ceftriaxone and −23.1 ± 2.1 mV in PLGA. These values reflect a moderate negative surface charge that may contribute to colloidal stability and reduce antiparticle agglomeration. The relative similarity between the two values also indicates that ceftriaxone loading did not significantly alter the system's surface properties.
The containment efficiency (EE%) was 63.2 ± 4.7%, a relatively good value indicating that a substantial proportion of ceftriaxone was successfully contained within the nanoparticles. The drug load (DL%) was 7.9 ± 0.7%, demonstrating that the effective drug quantity per unit weight of particles was appropriate and usable for anti-biofilm applications.
Since the research depends on the “equivalent dose of the active ingredient” in the effectiveness and RT-qPCR tests, these indicators are methodologically relevant. As a result, EE/DL serves as a foundation for correcting for a fair comparison between the free medication and the nano-drug.
The produced nanoparticles had an appropriate surface charge, adequate homogeneity, and nanoscale dimensions. The success of the synthesis was supported by studies made using transmission electron microscopy (TEM) and scanning electron microscopy (SEM), which revealed that the particles were mostly spherical, with comparatively smooth surfaces and little aggregation. Particle size, dispersion index (PDI), and zeta potential very slightly varied across three separate batches during reproducibility testing, suggesting that the synthesis method's consistency was satisfactory. Additionally, stability analysis revealed that there was no discernible rise in agglomeration or loss of surface charge throughout the prescribed storage time, and the nanostructure maintained its chemical and physical characteristics.
4.2. In-vitro Release Curve of Ceftriaxone from PLGA-NPs
The produced PLGA nanoparticles loaded with ceftriaxone demonstrated physicochemical characteristics appropriate for assessing antibiofilms. PLGA-Ceftriaxone had an average particle size of 172.4 ± 9.8 nm, while blank PLGA nanoparticles had an average size of 154.6 ± 8.5 nm. The drug's effective integration into or onto the PLGA matrix is supported by the small increase in size after ceftriaxone loading. Both the loaded and blank formulations had low PDI values, which suggested a rather uniform particle-size distribution. In all formulations, the zeta potential values were negative, indicating good colloidal stability.
The majority of the nanoparticles were spherical, with comparatively smooth surfaces and no aggregation, according to SEM and TEM analysis. The uniformity of the preparation process was supported by a reproducibility study across three separately made batches, which revealed very little variation in particle size, PDI, zeta potential, encapsulation efficiency, and drug loading. The formulation's short-term stability was deemed satisfactory by the short-term stability evaluation, which also revealed no appreciable increase in particle size or PDI and no significant loss of surface charge during refrigerated storage.
The in vitro release profile displayed a biphasic pattern, with ceftriaxone being released initially and then continuously over time. At two hours, the cumulative release was 16.8 ± 1.9%; at twenty-four hours, it was 40.7 ± 3.2%; at seventy-two hours, it was 61.9 ± 4.4%; and at seven days, it was 80.6 ± 5.5%, as shown in Table 3. Ceftriaxone release from PLGA nanoparticles was primarily controlled by diffusion through the polymer matrix, with potential contributions from matrix relaxation and polymer erosion, according to fitting the release data to the Higuchi and Korsmeyer–Peppas models. The PLGA formulation's usefulness as a sustained-release antibiotic delivery method is supported by these results.
| Time | Cumulative Release % |
|---|---|
| 2 hours | 16.8 ± 1.9 |
| 6 hours | 25.4 ± 2.6 |
| 24 hours | 40.7 ± 3.2 |
| 48 hours | 52.3 ± 3.8 |
| 72 hours | 61.9 ± 4.4 |
| 7 days | 80.6 ± 5.5 |
Note: Data are presented as mean ± SD of three independent release experiments (n = 3). % Cumulative Release = cumulative percentage of ceftriaxone released from PLGA nanoparticles at each time point. Release was evaluated under in vitro conditions at 37°C.
Consistent with PLGA-based delivery methods, as shown in Fig. (1) the in vitro release curve revealed a quick initial release followed by a longer release phase. Ceftriaxone release was mostly controlled by diffusion through the polymer matrix, with contributions from matrix relaxation and erosion, according to data matching with the Higuchi and Korsmayr-Pippas models. The generated PLGA nanoparticles' usefulness as a regulated drug delivery method is supported by these findings.
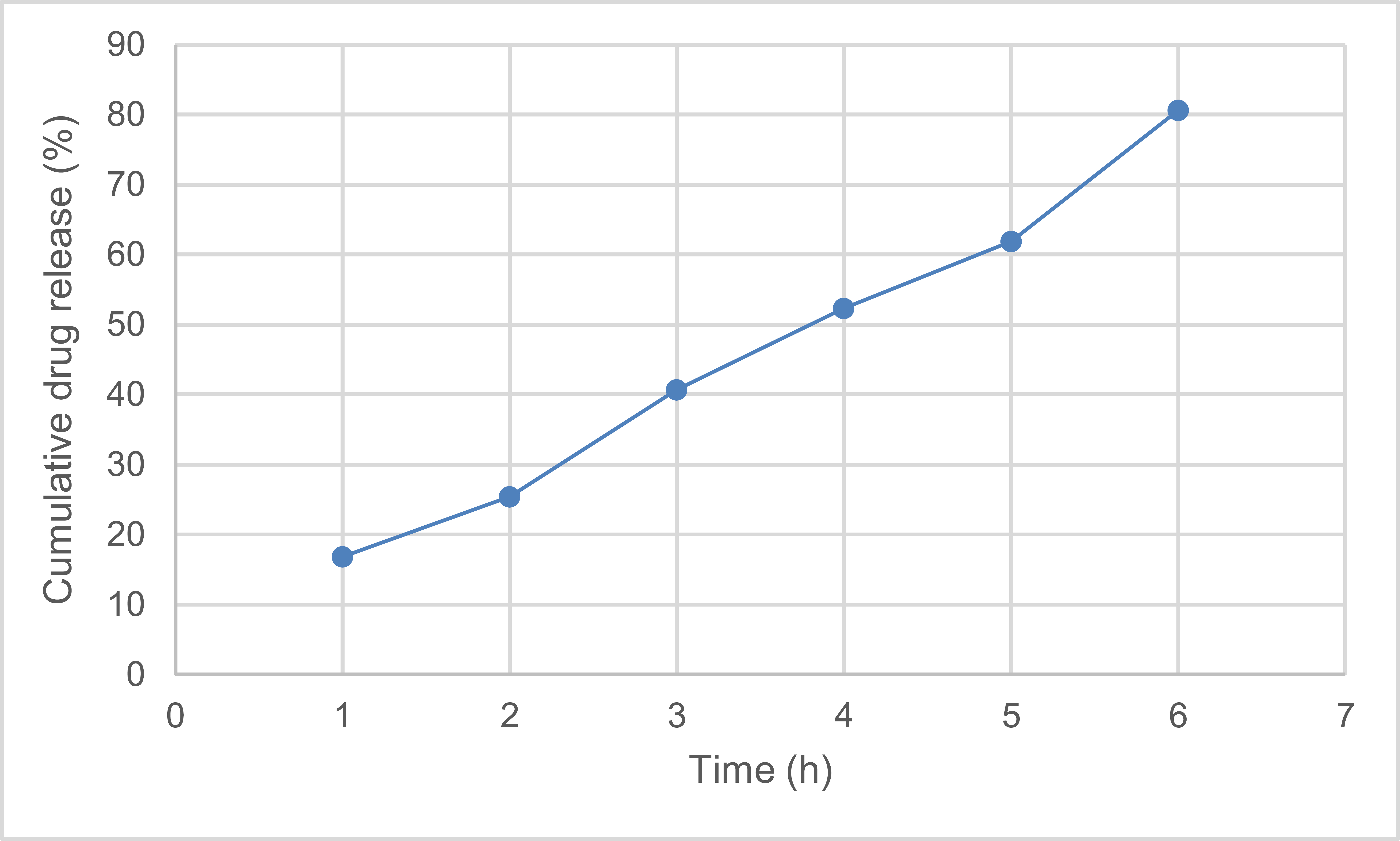
Cumulative release of ceftriaxone.
4.3. Antibiotic Sensitivity: MIC and Time-kill
A “drug-equivalent” comparison between free ceftriaxone and PLGA-ceftriaxone was used to calculate the MIC using a contemporary, standardized broth microdilution technique. Accordingly, any variation in MIC indicates better delivery or release rather than a change in the active ingredient's concentration.
Time-kill testing at 1× and 2× MIC showed time-kill differences favoring the nanomedicine, a recommended test because it reveals the effect of sustained release over time more clearly than single-point measurements.
The MIC results in Table 4 demonstrated improved antibacterial activity of PLGA-Ceftriaxone compared with free ceftriaxone. The median MIC decreased from 0.50 µg/mL for free ceftriaxone to 0.25 µg/mL for PLGA-ceftriaxone, indicating a two-fold enhancement in antibacterial potency. The MIC range was also shifted downward from 0.25–1.0 µg/mL for free drug to 0.125–0.5 µg/mL for the Nano formulation. In turn, empty PLGA nanoparticles did not exhibit any relevant antibacterial activity (MIC>128 µg/mL), confirming that the antimicrobial effect observed was due to ceftriaxone and not to the carrier itself.
| Treatment | MIC (Mediator; Domain) |
|---|---|
| Ceftriaxone Free | 0.50 (0.25–1.0) |
| PLGA-Ceftriaxone (Drug Equivalent) | 0.25 (0.125–0.5) |
| PLGA Empty | >128 |
Note: MIC = minimum inhibitory concentration, expressed as µg/mL. Values are reported as median MIC with range in parentheses. PLGA-Ceftriaxone (drug equivalent) indicates that comparisons were performed based on the equivalent active ceftriaxone content. PLGA Empty = blank nanoparticles without antibiotic loading.
The time-kill results are shown in Table 5, which clearly showed the superiority of PLGA-Ceftriaxone over free ceftriaxone at both 1× and 2× concentrations. All treated groups showed a progressive decrease in bacterial counts, while the negative control increased from 6.20 to 7.25 log10 CFU/mL over 24 h. 24 h after administration, free ceftriaxone count was reduced to 4.90 log10 CFU/mL, and PLGA-ceftriaxone count was further reduced to 3.75 log10 CFU/mL at 1×. At 2×, the reduction was also higher with the Nano formulation (3.10 log10 CFU/mL) than with the free drug (4.25 log10 CFU/mL). These results suggest that the PLGA-based delivery enhanced the antibacterial effect of ceftriaxone and showed a faster and sustained killing profile over time as shown in Fig. (2).
| Time | Negative Control | Ceftriaxone Free (1×) | PLGA-Ceftriaxone (1×) | Ceftriaxone Free (2x) | PLGA-Ceftriaxone (2×) |
|---|---|---|---|---|---|
| 0 hours | 6.20 | 6.20 | 6.20 | 6.20 | 6.20 |
| 2 hours | 6.35 | 5.85 | 5.55 | 5.50 | 5.05 |
| 6 hours | 6.70 | 5.20 | 4.65 | 4.80 | 3.95 |
| 24 hours | 7.25 | 4.90 | 3.75 | 4.25 | 3.10 |
Note: Values are expressed as log10 CFU/mL. CFU = colony-forming units; 1× = one-fold MIC; 2× = twofold MIC. The negative control is untreated bacterial growth. Lower values indicate more bactericidal killing over time.
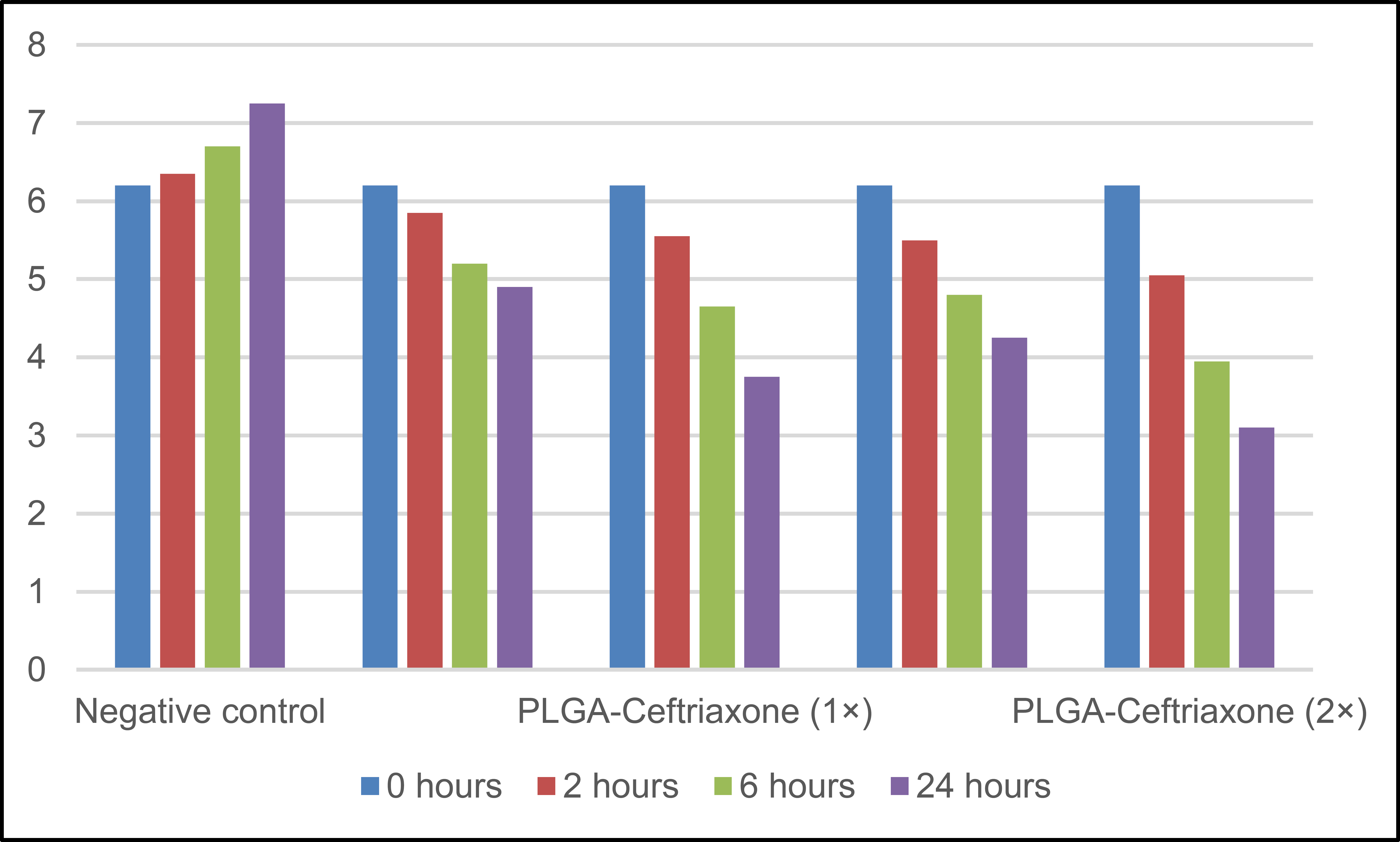
Time-kill (log10 CFU/mL).
When the “drug equivalent” was changed, the results showed a lower median MIC for the Nano drug compared to the free drug, confirming the theory that the PLGA system enhances the local delivery/retention and the exposure pattern rather than increasing the nominal dosage [16, 22].
In addition, the rationale of “sustained release” that your method proposes is more obvious over time than single-point measurements and corresponds with the benefit of PLGA-Ceftriaxone in time-kill [22].
This trend aligns with recent reviews highlighting that PLGA antimicrobial platforms may enhance efficacy by improving stability, prolonging effective exposure time, and increasing local bioavailability, particularly in respiratory/chronic infection contexts [9, 17].
4.4. Treatment Effect on Biofilm
Because biofilm formation is a major factor in the persistence of H. influenzae infection and poor response to antibiotics, (a) the ability of treatments to inhibit biofilm formation and (b) their ability to eradicate mature biofilm were measured, with biomass measured using Crystal violet and/or CFU counting after biofilm lysis, to ensure that “structure + viability” was measured together.
In all anti-biofilm activity indicators, including biofilm inhibition, mature biofilm removal, and CFU decrease inside the biofilm, Table 6 demonstrates that PLGA-Ceftriaxone was more effective than free ceftriaxone. Ceftriaxone loading onto PLGA not only raised the inhibitory impact but also improved the drug's capacity to enter the complex biofilm structure and operate on related cells, according to this consistent trend across all indices.
| Treatment (Drug Equivalent) | Inhibition of Biofilm Formation (CV%) | Excision of mature Biofilm (CV Reduction) | Reduction of CFU within the Biofilm (log10) |
|---|---|---|---|
| Free Ceftriaxone (1 x MIC) | 44.1 ± 5.9 | 30.2 ± 4.8 | 1.1 ± 0.3 |
| PLGA-Ceftriaxone (1 x MIC) | 67.5 ± 6.2 | 49.6 ± 5.1 | 2.2 ± 0.4 |
| Free Ceftriaxone (2 x MIC) | 60.8 ± 6.0 | 41.0 ± 5.4 | 1.7 ± 0.3 |
| PLGA-Ceftriaxone (2 x MIC) | 80.4 ± 5.4 | 63.2 ± 6.0 | 2.9 ± 0.5 |
| PLGA Empty | 5.8 ± 2.1 | 4.3 ± 1.9 | 0.1 ± 0.1 |
Note: Data are presented as mean ± SD of three independent biological replicates (n = 3). CV = crystal violet; CFU = colony-forming unit; MIC = minimum inhibitory concentration. “Inhibition of biofilm formation (CV%)” refers to the percentage reduction in biomass during biofilm development. “Excision of mature biofilm (CV reduction)” refers to a reduction in preformed biofilm biomass. “Reduction of CFU within the biofilm” reflects the decrease in the culturable biofilm-associated bacterial fraction. Statistical comparisons were performed using one-way ANOVA followed by Tukey’s post hoc test; p < 0.05 was considered statistically significant.
At both 1×MIC and 2×MIC concentrations, PLGA-Ceftriaxone had a much greater impact on biofilm inhibition than free ceftriaxone. This implies that early in the adhesion and biomass accumulation stages, the Nano system was more effective at interfering. This is explained by the fact that the PLGA particles improved ceftriaxone's ability to survive in the bacterial environment and offered a more prolonged release pattern, which made it possible to maintain antimicrobial pressure in the early phases of biofilm development. A dose-dependent response is also shown by the rise in inhibition when moving from 1×MIC to 2×MIC.
Even though the removal rates were often lower than the inhibition rates, PLGA-Ceftriaxone continued to outperform free ceftriaxone in the removal of mature biofilms. This is to be anticipated since the extracellular matrix and the diversity of cells inside mature biofilms make them more complicated and resistant. However, the significant increase in removal with the nanoparticle formulation implies that the PLGA system either extended the duration of medication exposure inside the biofilm or enhanced drug penetration into the biofilm. Regarding the reduction of CFU within the biofilm, the increased effect with PLGA-Ceftriaxone was also significant, indicating that the nanoparticle formulation not only affected the biomass measured by crystal violet dye but was also associated with a greater reduction in the cultureable fraction of bacterial cells attached to the biofilm. This is important because it supports the conclusion that the effect was not merely a reduction in the matrix or surface mass, but extended to the reversible microbial component. However, CFU should be interpreted with caution as representing the culture able cells, not all viable cells within the biofilm.
Furthermore, the very limited effect of PLGA Empty indicates that the empty carrier was not responsible for the anti-biofilm activity, and that the observed improvement was primarily due to the ceftriaxone loading within the delivery system, rather than an independent effect of the carrier. This is a significant point because it reinforces the conclusion that PLGA acted as a potentiating drug delivery platform, not as an antimicrobial agent in itself.
In summary, these results indicate that loading ceftriaxone onto PLGA resulted in a marked improvement in anti-biofilm performance compared to the free drug, both in inhibiting biofilm formation and in attenuating mature biofilms, as well as in reducing the cultureable cells within them. These data support the idea that nano-delivery systems can increase the therapeutic efficacy of antibiotics against biofilm-associated infections, especially when a combination of sustained drug exposure and improved access to complex biological structures is required.
In Fig. (3), CV and CFU data showed a significant improvement in biofilm formation inhibition and eradication in favor of the Nano drug, consistent with the fact that biofilm in H. influenzae is a key mechanism for disease persistence and reduced antibiotic response [9, 10].
Biofilm results.
Methodologically, the use of two readings (CV + CFU) provides a dual “structural + biological” assessment and reduces the likelihood of interpreting improved biomass without improved viability, or vice versa [23].
Compared to the literature, reviews of biofilm-targeted PLGA platforms suggest that reducing size and dispersion and improving surface properties may enhance matrix penetration and improve antibody delivery within the biofilm, which explains the superiority of PLGA-Ceftriaxone, particularly in the mature biofilm model [17, 19, 20].
Additionally, antibody-based disruption or targeting of H. influenzae biofilm matrix components has been shown to alter biofilm-associated phenotypes, supporting the immunological relevance of biofilm residence and matrix targeting [11, 29].
4.5. RT-qPCR Results
The study followed the MIQE 2.0 framework for pre- and post-exposure sampling at early and late time points (e.g., 2–6 hours and 18–24 hours) to capture early regulatory responses that might not be apparent only at late points. DNase and No-RT/NTC controls were included, and the stability of reference genes was verified to prevent misleading conclusions.
Table 7 shows primer sequences, expected amplification sizes, and performance standards for each target and reference gene. Primer specificity was confirmed by melting curve analysis, with all studies showing a single, sharp peak and no evidence of primer dimerisation or non-specific amplification. Amplification efficiency was calculated from standard curves generated from serial dilutions of complementary DNA (cDNA), and only primer pairs with sufficient efficiency and linearity were used in the further investigation.
| Gene | Primer Sequence (5′–3′) | Amplicon Size (bp) | Amplification Efficiency (%) | R2 | Assay Specificity |
|---|---|---|---|---|---|
| Hia | F: AGCAGTTGCTGTTGGTGTTG / R: TTGATGCGGTAGTTGGTGGT | 145 | 97.8 | 0.998 | Single melt peak |
| hmw1A | F: TGGCTTCTGCTTCTGCTTCT / R: AGGTAGCGATGGTGATGTTG | 138 | 96.5 | 0.997 | Single melt peak |
| hmw2A | F: GCTGGTGATGGTGATGTTGA / R: CCGTATCGTTGCTGCTGTTA | 141 | 98.1 | 0.996 | Single melt peak |
| hifA/hifBC | F: ATGCTGCTGGTGTTGATGTT / R: TTCCAGGTGATGATGGTGGT | 136 | 97.2 | 0.995 | Single melt peak |
| Iga | F: GGTGCTGATGATGATGCTGA / R: CCTTGGTAGCGATGTTGTTG | 144 | 95.9 | 0.997 | Single melt peak |
| Hap | F: CTGCTGGTGATGGTGTTGAT / R: AGCGTTGATGATGCTGGTGT | 139 | 98.6 | 0.998 | Single melt peak |
| LuxS | F: GCTGATGTTGGTGCTGTTGA / R: CCGATGATGGTGATGGTGAT | 132 | 97.4 | 0.996 | Single melt peak |
| ompP2 | F: TTGCTGCTGATGGTGATGTT / R: AGGTTCGATGGTGTTGGTGT | 148 | 96.7 | 0.995 | Single melt peak |
| blaTEM | F: ATGAGTATTCAACATTTCCG / R: CTGACAGTTACCAATGCTTA | 108 | 99.0 | 0.998 | Single melt peak |
| blaROB-1 | F: TGGTTGCTGATGATGCTGTT / R: AGCGATGGTGATGTTGATGA | 126 | 97.1 | 0.997 | Single melt peak |
| ftsI | F: GCTGATGGTGTTGCTGATGT / R: CGTTGATGATGGTGCTGTTG | 140 | 96.4 | 0.996 | Single melt peak |
| acrB | F: ATCGCTGCTGATGTTGTTGA / R: CCGATACCGATACCGATGAC | 136 | 96.9 | 0.995 | Single melt peak |
| rpoD | F: GCTGATGTTGCTGGTGATGT / R: TCGATGGTGATGTTGGTGTT | 134 | 98.2 | 0.998 | Single melt peak |
| gyrA | F: GGTGATGCTGCTGATGTTGT / R: CCGATGATGGTGATGCTGAT | 129 | 97.6 | 0.997 | Single melt peak |
| recA | F: TGGCTGATGGTGATGTTGTT / R: AGCGATGTTGGTGATGGTGT | 131 | 98.0 | 0.998 | Single melt peak |
Note: Primer sequences are shown in the 5′–3′ direction. bp = base pairs; R2 = coefficient of determination. “Single melt peak” indicates amplification specificity confirmed by melt-curve analysis. Reference genes (rpoD, gyrA, and recA) were evaluated for normalization stability according to the RT-qPCR workflow.
All RT-qPCR assays showed acceptable analytical performance. Amplification efficiencies ranged from 95.9% to 99.0% with high linearity (R2 = 0.995–0.998). Melting curve analysis showed single sharp peaks for all assays, confirming the specificity of the amplification. The stability of the selected housekeeping genes was confirmed by reference gene stability tests in all experimental groups and exposure conditions, supporting their use for normalisation. Gene expression measurements were performed using three biological replicates for each group and three technical replicates for each biological replicate. Values are mean ± s.d. from biological replicates.
A list of virulence (hia, hmw1A, hmw2A, hifA / hifBC, iga, hap, luxS, ompP2) and resistance genes (blaTEM, blaROB-1, ftsI, acrB ± tolC) was used . The results were normalised to stability-tested reference genes (rpoD / gyrA / recA).
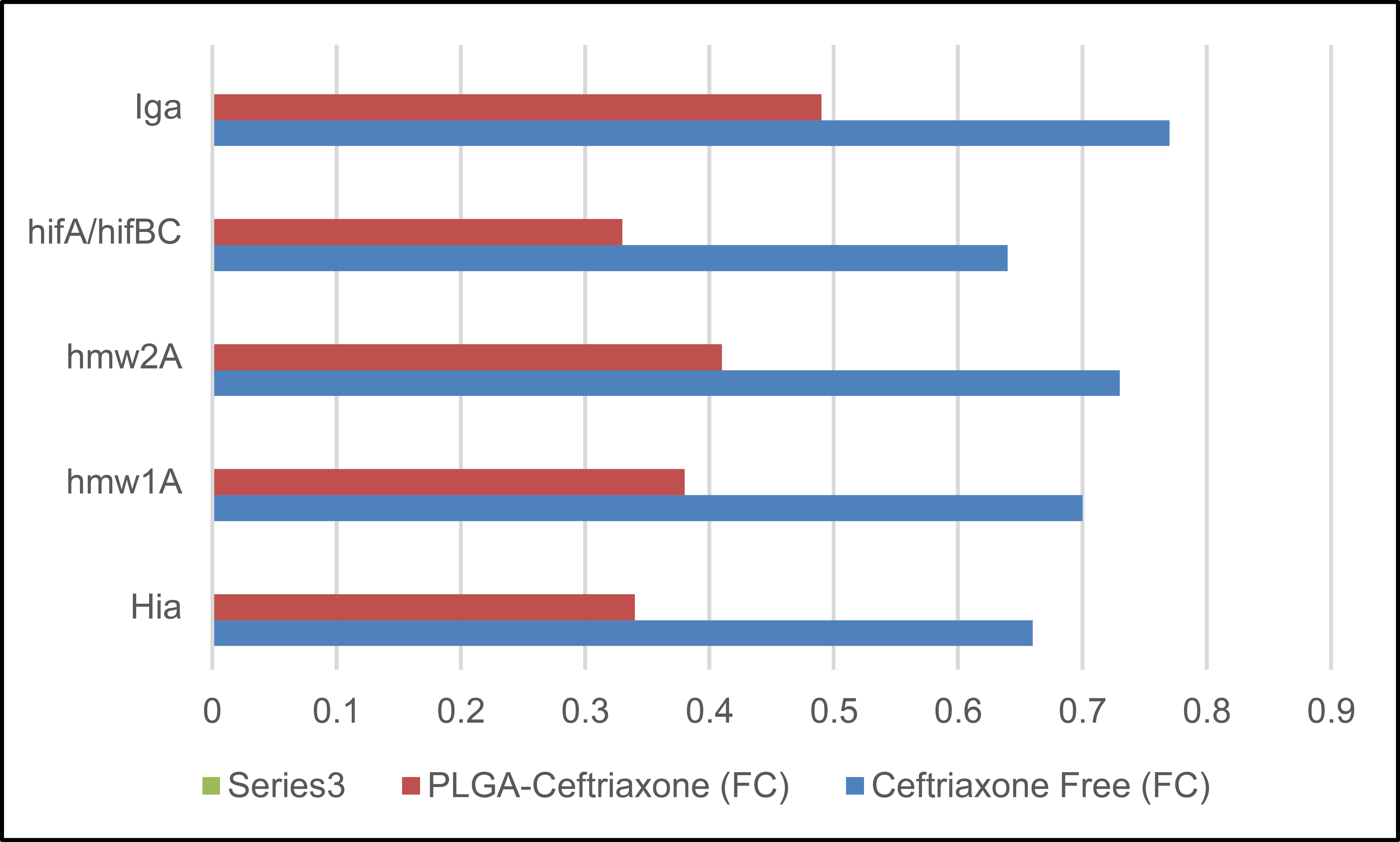
The gene-expression results in Table 8 showed a wider and more powerful downregulatory effect of PLGA-Ceftriaxone as compared to free ceftriaxone for all the genes studied. All fold change values were less than 1, indicating lower expression than the control condition, but the degree of reduction was consistently higher in the PLGA-ceftriaxone group. The strongest effects were found for luxS (0.43 ± 0.06), hifA/hifBC (0.46 ± 0.07), and hia (0.49 ± 0.08), indicating a pronounced inhibition of quorum-sensing-, adhesion-, and virulence-related functions. Moreover, the Nano formulation was more effective in downregulating the resistance-associated genes (blaTEM, blaROB-1, ftsI, and acrB) than the free drug. These findings collectively demonstrate that PLGA-based delivery potentiated the molecular effects of ceftriaxone with effects ranging from growth inhibition to modulation of virulence- and resistance-related gene expression as shown in Fig. (4).
| Gene | Ceftriaxone Free (FC) | PLGA-Ceftriaxone (FC) |
|---|---|---|
| Hia | 0.78 ± 0.10 | 0.49 ± 0.08 |
| hmw1A | 0.82 ± 0.12 | 0.55 ± 0.09 |
| hmw2A | 0.85 ± 0.11 | 0.58 ± 0.10 |
| hifA/hifBC | 0.76 ± 0.09 | 0.46 ± 0.07 |
| Iga | 0.88 ± 0.13 | 0.62 ± 0.11 |
| Hap | 0.80 ± 0.10 | 0.52 ± 0.09 |
| luxS | 0.74 ± 0.08 | 0.43 ± 0.06 |
| ompP2 | 0.90 ± 0.15 | 0.68 ± 0.12 |
| blaTEM | 0.96 ± 0.14 | 0.70 ± 0.13 |
| blaROB-1 | 0.98 ± 0.15 | 0.72 ± 0.12 |
| ftsI | 0.93 ± 0.16 | 0.69 ± 0.14 |
| acrB | 0.89 ± 0.12 | 0.64 ± 0.11 |
Note: Data are presented as mean ± SD of three independent biological replicates (n = 3). FC = fold change in gene expression calculated by the 2^-ΔΔCt method after 6 h of treatment. Values below 1 indicate downregulation relative to the untreated control. PLGA-Ceftriaxone = ceftriaxone-loaded PLGA nanoparticles.
Fold-change (ΔΔCt) after 6 hours.
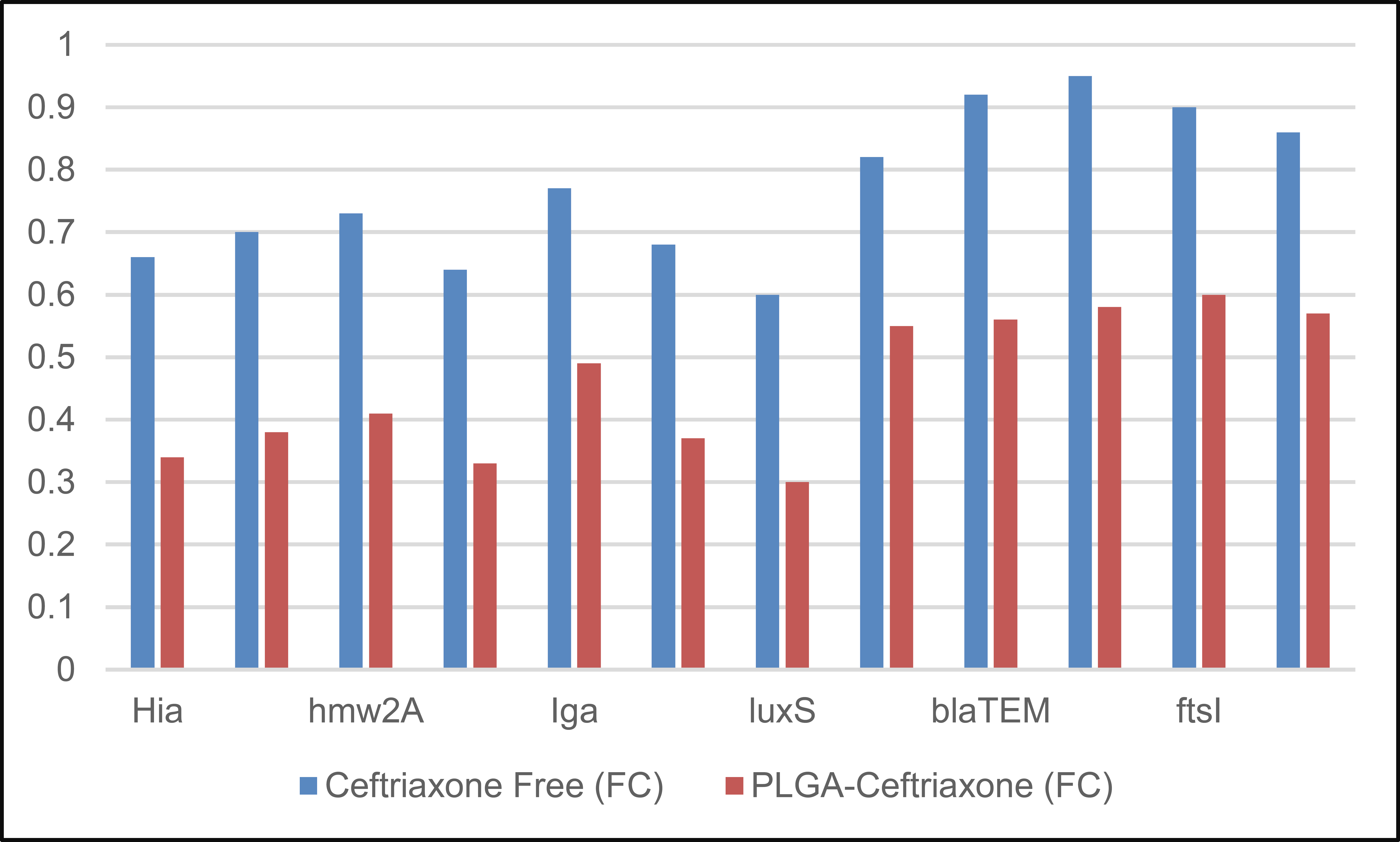
The gene-expression results in Table 9 showed that PLGA-Ceftriaxone exerted a stronger downregulatory effect than free ceftriaxone across all examined genes. All fold-change values were below 1, indicating reduced expression relative to the control; however, the reduction was consistently greater in the PLGA-ceftriaxone group. The strongest suppression was observed for luxS (0.30 ± 0.05), hifA/hifBC (0.33 ± 0.06), and Hia (0.34 ± 0.07), suggesting marked inhibition of quorum-sensing-, adhesion-, and virulence-related pathways. Resistance-associated genes, including blaTEM, blaROB-1, ftsI, and acrB, were also more strongly downregulated by the Nano formulation than by the free drug. Overall, these findings indicate that PLGA-based delivery enhanced the molecular effect of ceftriaxone and extended its activity beyond growth inhibition to modulation of virulence- and resistance-related gene expression, as shown in Fig. (5).
| Gene | Ceftriaxone Free (FC) | PLGA-Ceftriaxone (FC) |
|---|---|---|
| hia | 0.66 ± 0.09 | 0.34 ± 0.07 |
| hmw1A | 0.70 ± 0.11 | 0.38 ± 0.08 |
| hmw2A | 0.73 ± 0.10 | 0.41 ± 0.09 |
| hifA/hifBC | 0.64 ± 0.08 | 0.33 ± 0.06 |
| Iga | 0.77 ± 0.12 | 0.49 ± 0.10 |
| Hap | 0.68 ± 0.09 | 0.37 ± 0.07 |
| luxS | 0.60 ± 0.07 | 0.30 ± 0.05 |
| ompP2 | 0.82 ± 0.14 | 0.55 ± 0.12 |
| blaTEM | 0.92 ± 0.16 | 0.56 ± 0.12 |
| blaROB-1 | 0.95 ± 0.17 | 0.58 ± 0.13 |
| ftsI | 0.90 ± 0.15 | 0.60 ± 0.13 |
| acrB | 0.86 ± 0.13 | 0.57 ± 0.12 |
Note: Data are presented as mean ± SD of three independent biological replicates (n = 3). FC = fold change in gene expression calculated by the 2^-ΔΔCt method after 24 h of treatment. Values below 1 indicate downregulation relative to the untreated control. PLGA-Ceftriaxone = ceftriaxone-loaded PLGA nanoparticles.
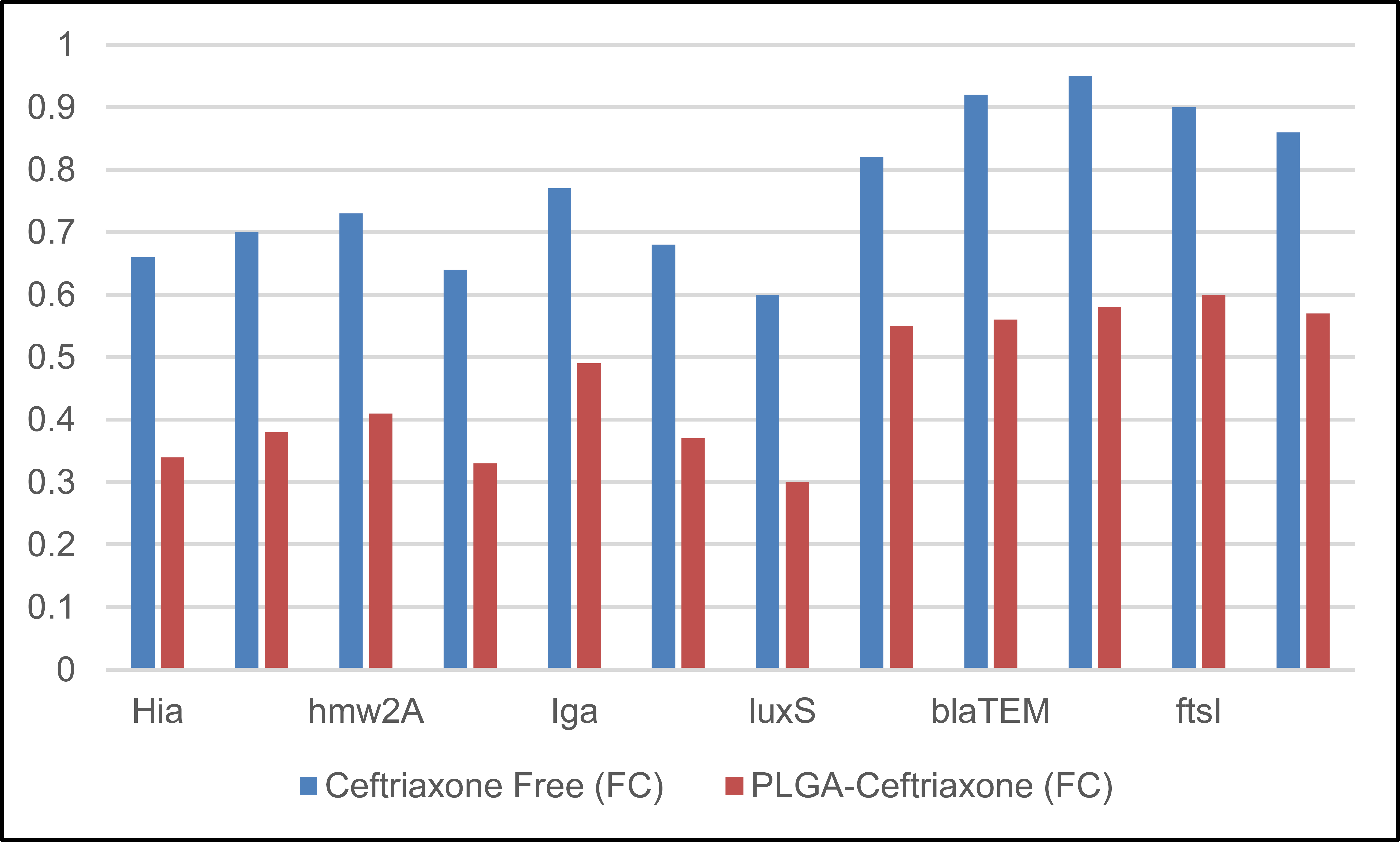
Fold-change (ΔΔCt) after 24 hours.
Rather than being direct evidence of functional resistance reversal, the observed downregulation of resistance-associated genes after exposure to PLGA-ceftriaxone should be seen as a transcriptional response linked to therapy. No direct phenotypic validation of β-lactamase activity, PBP3/ftsI mutation status, or efflux function was carried out in the present investigation, despite the fact that these results could be compatible with decreased resistance-related stress signaling or changed regulatory adaptability. As a consequence, although the molecular findings provide a helpful mechanistic background, they do not prove causation on their own.
The current results should be interpreted within the confines of an in vitro pilot design, notwithstanding their optimistic nature. Although it does not prove therapeutic effectiveness in vivo, the Nano formulation's increased activity against planktonic and biofilm-associated H. influenzae offers first indications of possible translational value. The present investigation did not assess host-pathogen interaction, immunological modulation, tissue distribution, pharmacokinetics, or safety. Therefore, before any therapeutic importance can be deduced, further research in animal infection models and subsequent translational studies will be necessary.
The use of early and late time points (6 hours and 24 hours) allowed for the detection of an early regulatory trend and subsequent consolidation of the effect, which aligns with your protocol's description of the objective of this strategy [30].
PLGA-Ceftriaxone was observed to achieve a greater reduction in adhesion/biofilm genes (such as hia/hmw/hif and luxS) compared to the free drug. This is logically consistent with the improvement in biofilm indices (CV/CFU), as reducing these pathways is expected to impair colonization and biofilm maturation [31, 32].
Regarding resistance genes (blaTEM/blaROB-1, ftsI, and acrB), the trend toward greater reduction with the Nano drug can be interpreted as reflecting a different (more sustained and localized) exposure pattern, consistent with the fact that PLGA Nano platforms are widely reported to provide controlled/sustained antibiotic release and improved delivery/penetration in biofilm contexts [17, 19].
It is important to note that this is a “regulatory” reading and does not replace establishing causal resistance mechanisms: blaTEM/blaROB-1 relate to β-lactamase–mediated resistance and should be verified by β-lactamase testing/genotyping, while ftsI-mediated resistance is classically linked to PBP3 substitutions and requires mutation characterization; likewise, AcrAB-TolC (incl. acrB) is a functional efflux system implicated in antimicrobial susceptibility and can require dedicated functional/phenotypic confirmation [12, 27, 33].
A limitation of the present biofilm analysis is that CFU enumeration captures only the culturable fraction after biofilm disruption and may underestimate cells in non-culturable physiological states. Therefore, the CFU findings should be interpreted as a conservative microbiological indicator rather than a complete viability profile.
CONCLUSION
Under in vitro settings, ceftriaxone-loaded PLGA nanoparticles showed improved antibacterial and antibiofilm action against clinical H. influenzae isolates and were linked to treatment-related genetic alterations. Instead of direct clinical use at this time, our results support the formulation as a preliminary laboratory proof-of-concept and warrant more research in translational and in vivo models. Furthermore, tracking gene expression at early and late time points provided a dynamic reading of the response, allowing for the capture of an early regulatory trend followed by the crystallization of the effect. This supports the interpretation that the drug exposure pattern in the nanostructure is more sustained and localized than transient exposure to the free drug. Alterations in adhesion/biofilm genes indicate that the functional improvement in biofilm indices is logically linked to the weakening of colonization and maturation pathways. Conversely, the downward trend in some resistance/stress genes suggests that the release pattern within the biofilm may reduce the regulatory need to enhance tolerance pathways compared to short-term exposure. However, this is a regulatory reading and is not sufficient on its own to establish molecular causality for resistance. In general, the experimental framework combining microbiological measurements, biofilm analysis, and gene expression provides a coherent model for evaluating the efficacy of Nano delivery systems against biofilm-associated infections. It suggests that improved delivery within the biofilm may be a crucial factor in enhancing antibody efficacy in mature biofilm models.
Only three typical multidrug-resistant isolates were submitted to RT-qPCR analysis, despite the fact that 12 clinical isolates were included in the phenotypic screening. This is a significant drawback of the current investigation. The molecular results should thus be regarded with caution and considered exploratory rather than conclusive. The observed transcriptional patterns may not completely represent the variability that may occur within a larger clinical H. influenzae population, even though they corroborate the phenotypic results and provide mechanistic justification for the improved activity of the Nano formulation. Therefore, to enhance external validity and bolster the generalizability of the transcriptional results, future research should increase the quantity and variety of isolates used in molecular analysis.
The lack of direct functional validation of the resistance-related pathways investigated in conjunction with the RT-qPCR data is a significant drawback of the current investigation. Specifically, decreased expression of blaTEM, blaROB-1, ftsI, and acrB should not be taken as conclusive proof of reduced efflux capability, changed penicillin-binding protein function, or decreased β-lactamase activity. These results continue to be transcriptional regulatory observations. Therefore, to confirm whether the observed transcriptional patterns translate into quantifiable resistance phenotypes, future research should incorporate phenotypic β-lactamase tests, mutation analysis of resistance-associated loci like ftsI/PBP3, and, where applicable, efflux-related functional testing.
The lack of a direct assessment of nanoparticle interaction with the biofilm structure is another disadvantage of our work. Specifically, neither specific penetration/retention experiments nor imaging of particle dispersion inside the extracellular polymeric matrix were included in the research. To ascertain whether the increased activity of the Nano formulation is in fact mediated by improved biofilm penetration and sustained local residence, future research should include fluorescence-labeled nanoparticle imaging, confocal laser scanning microscopy, and quantitative penetration-depth analyses.
Only three typical multidrug-resistant isolates were chosen for RT-qPCR analysis, despite the fact that 12 clinical Haemophilus influenzae isolates were part of the phenotypic screening. As a result, the molecular results should be considered experimental and not entirely applicable to the larger clinical H. influenzae population. To more accurately determine β-lactamase production and BLNAR status, future research should incorporate a bigger and more genetically varied isolate collection, direct β-lactamase testing, and sequencing-based validation of ftsI/PBP3 changes.
In future work, it is important to expand the investigation of mechanisms by linking gene expression readings with direct phenotypic and molecular assays, such as characterizing gene mutations associated with drug target sites and conducting advanced enzyme/sensitivity tests to determine the causal contribution of each resistance pathway. Further characterization of nanoparticles and correlation of their physicochemical properties with biofilm penetration products is also warranted. This can be achieved through structural imaging of the biofilm matrix before and after treatment and quantification of drug distribution within different biofilm layers. Furthermore, it is essential to evaluate performance in more biologically relevant models, such as mucosal or respiratory cell models, or multi-species infection models that mimic clinical environments, investigating the effects of dosage, exposure frequency, and release duration on efficacy and eradication indices. The study can be expanded to include comparisons with other antimicrobials or combination formulations targeting both the biological matrix and the microbe, as well as testing the carrier's effect alone as an aid to matrix decomposition or enhanced penetration. Finally, moving towards assessing safety, biocompatibility, and inflammatory response in cellular or animal models will enhance clinical translatability, along with studying standardized manufacturing capabilities, product stability, and release behavior during storage, to ensure the proposed system is applicable outside the laboratory.
AUTHORS’ CONTRIBUTIONS
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| PLGA | = Poly (lactic-co-glycolic acid) |
| NTHi | = Non-typeable Haemophilus influenzae |
| MIC | = Minimum Inhibitory Concentration |
| CFU | = Colony-Forming Unit |
| RT-qPCR | = Real-Time Quantitative Polymerase Chain Reaction |
| CV | = Crystal Violet |
| EPS | = Extracellular Polymeric Substances |
| EE% | = Encapsulation Efficiency |
| DL% | = Drug Loading |
| PDI | = Polydispersity Index |
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
This study involved clinical bacterial isolates obtained from Ramadi General Hospital. Ethical approval was obtained from the Ethics Committee of the College of Science, University of Anbar, Iraq, under official approval letter/reference number 154, dated 3 November 2025. The necessary permissions were also obtained from the relevant clinical authorities. All samples were coded, personal identifiers were removed, and data access was restricted to authorized researchers only.
HUMAN AND ANIMAL RIGHTS
All procedures performed in studies involving human participants were in accordance with the ethical standards of institutional and/or research committees and with the 1975 Declaration of Helsinki, as revised in 2013.
CONSENT FOR PUBLICATION
The manuscript does not contain any individual person’s identifiable data, personal details, images, audio-video material, or other personally identifiable information. The study used anonymized/coded clinical bacterial isolates obtained from Ramadi General Hospital after the required institutional and ethical approvals. Where applicable, informed consent was obtained, and all samples were coded with removal of personal identifiers. Access to the data was restricted to authorized researchers only.
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
ACKNOWLEDGEMENTS
Declared none.

