All published articles of this journal are available on ScienceDirect.

Development of a Recombinant Live Attenuated Influenza Vaccine Virus Expressing Pneumococcal Surface Antigen A as a Strategy for Combined Protection Against Influenza and Bacterial Coinfection Caused by Streptococcus pneumoniae
Authors Info & Affiliations
Abstract
Introduction
Bacterial superinfection with Streptococcus pneumoniae following the influenza virus infection complicates the course of the disease and is a major cause of mortality during influenza virus epidemics. The effectiveness of licensed polysaccharide vaccines is limited by the serotypes included in the vaccine and possible immune tolerance during revaccination. Pneumococcal surface protein A (PspA), which includes conserved regions and has at least two functions in pathogen virulence, is considered a promising target for the development of new-generation vaccines, including gene-engineering constructions. Therefore, the development of an influenza virus vector-based vaccine expressing conserved bacterial proteins seems to be a promising strategy for designing combined vaccines against influenza and bacterial pathogens.
Methods
A recombinant live attenuated influenza virus (LAIV) expressing PspA fragment in a modified hemagglutinin was rescued on the A/Leningrad/134/17/57 (H2N2) backbone. This recombinant virus was assessed for its growth characteristics in vitro, as well as for its immunogenicity and protective capacity, using a mouse model of influenza-bacterial coinfection.
Results
The rescued recombinant LAIV/HA+PspA virus was genetically stable after sequential passaging in embryonated chicken eggs and possessed an attenuated phenotype similar to the classical LAIV strain. The LAIV/HA+PspA bivalent vaccine-induced IgG antibodies specific to both influenza virus and S. pneumoniae and provided complete protection of vaccinated mice against lethal influenza infection, as well as a 40% survival rate for lethal homologous and heterologous influenza infection complicated by concomitant bacterial infection with S. pneumoniae.
Conclusion
The presented design of a recombinant influenza virus carrying immunogenic fragments of a bacterial pathogen can be considered a promising strategy for the combined protection of vaccinated individuals against influenza and its bacterial complications, and further in-depth studies of such recombinant viruses in preclinical studies are warranted.
1. INTRODUCTION
Streptococcus pneumoniae (pneumococcus) is a gram-positive bacteria with over 100 distinct serotypes, which is the most common cause of bacterial pneumonia in children. S. pneumoniae is the leading cause of morbidity and mortality from lower respiratory tract infections worldwide and results in more than 1 million deaths annually [1].
S. pneumoniae has been progressively developing resistance against penicillins, macrolides, and linco- samides (erythromycin, clindamycin etc.) [2, 3]. Moreover, with global pandemics, the threat from antibiotic-resistant infections increases significantly [4-6]. According to the CDC Antibiotic Resistance Threats Report (2019), S. pneumoniae is listed as a bacteria of concerning threat [7, 8]. Preventing infections through vaccination reduces the use of antibiotics, which are a major factor in the development of antibiotic resistance. After the 13-valent pneumococcal vaccine (PCV13) was introduced in 2010, resistance to penicillin decreased significantly [9-12]. However, the incidence of antibiotic-nonsusceptible invasive pneumococcal disease (IPD) caused by serotypes other than PCV13 has increased [13, 14].
The antigens in existing vaccines are pneumococcal polysaccharides, which have a number of limitations. First, polysaccharides induce T-independent antibodies, which decrease over 3-7 years. In this case, revaccination with polysaccharide vaccines may not be effective due to possible immune tolerance [15]. Nevertheless, poly- saccharides are slightly immunogenic for infants and children under 2 years of age [16, 17]. Second, the effectiveness of polysaccharide vaccines is limited by the serotypes included in the vaccine, whereas the serotypes that cause IPD vary geographically [18, 19]. Moreover, the replacement of invasive pneumococcal disease with serotypes not covered by pneumococcal conjugate vaccine has been observed [20, 21]. Third, the polysaccharide antigens must be conjugated to a protein for enhanced immunogenicity, which limits the number of serotypes that can be included in a vaccine due to technical complexity in conjugating, as well as due to the high cost [22].
An alternative strategy is to develop protein-based vaccines that will utilize immunogenic and highly conserved proteins presented on the bacterial cell surface. Pneumococcal surface protein A (PspA) is considered a promising candidate for protein vaccines [17, 22] because it includes conserved regions [23] and has at least two functions in pathogen virulence. PspA competitively binds to lactoferrin (hLf), an iron-binding glycoprotein prevalent in human mucosal secretions. Upon binding of PspA-hLf, iron is released, sufficient levels of which on the mucosal surface are required for bacterial growth [24]. In addition, the N-domain of the PspA protein is negatively charged and appears on the surface of the bacterial cell, preventing the interaction of the complement system with the bacterium.
Influenza A virus is associated with an increased risk of infection and transmission of pneumococcal infection. Co-infection with both pathogens leads to severe, life-threatening pneumonia in humans [25, 26]. In the 1918 influenza pandemic, most deaths were attributed to secondary bacterial pneumonia, with pneumococcus being the most common infectious agent [27]. In general, about 16% of critically ill patients with influenza have bacterial coinfection with S. pneumoniae [28]. During the 2009 pandemic of H1N1 influenza, S. pneumoniae was the most common concomitant pathogen isolated in patients with fatal H1N1 infection [29]. Secondary bacterial pneumonia leads to a critical condition in which treatment with antibiotics does not always give a positive result because the destruction of the wall of the gram-positive bacterium S. pneumoniae under the influence of such antibiotics leads to the release of pro-inflammatory bacterial components that exacerbate the course of infection [30, 31].
Vaccination with live attenuated influenza vaccines (LAIVs) was shown to significantly reduce bacterial growth during subsequent influenza infection in mice, suggesting that LAIVs may significantly reduce bacterial disease during influenza epidemics in humans [32]. In the current study, we developed a combined vaccine prototype by incorporating a fragment of the PspA gene into the hemagglutinin gene of a live attenuated influenza virus and assessed this recombinant LAIV virus for its growth characteristics in vitro, as well as for the immunogenicity and protective capacity using a mouse model of influenza-bacterial coinfection.
2. MATERIALS AND METHODS
2.1. Viruses, Bacteria, Plasmids, and Sera
A/Leningrad/134/17/57 (H2N2) (A/Len/17), a cold-adapted master donor virus (MDV) for Russian LAIV, was used as a backbone for the development of vector bivalent vaccine strain. Six Len/17 genes encoding internal proteins (PB2, PB1, PA, NP, M, and NS) were earlier cloned into dual-promoter plasmids [33]. A 6:2 LAIV virus carrying HA and NA genes of A/Anhui/1/2013 (H7N9) avian influenza virus and six remaining genes of Len/17 MDV was used as an LAIV backbone for the generation of a recombinant LAIV/HA+PspA vaccine candidate [34].
Two A/PR8/34-based reassortant influenza viruses were used in mouse challenge experiments: (i) A/Shanghai/2/2013(H7N9) CDC-RG virus (Sh/PR8) expressing HA and NA genes of A/Shanghai/2/2013 (H7N9) virus and (ii) A/Philippines/2/1982 (X-79) (H3N2) virus. The H7N9 reassortant virus was obtained from the CDC (Atlanta, GA, USA). The H3N2 virus was a kind gift from Professor Florian Krammer (Icahn School of Medicine at Mount Sinai, New York, NY, USA). All influenza viruses were propagated in eggs and stored in single-use aliquots at -70°C.
A plasmid DNA based on pET24a expression vector encoding a hybrid protein comprising fragments of pneumococcal PspA protein (160-262 aa; GenBank EHD89 266.1 pneumococcal surface protein A (S. pneumoniae), Spr1895 (89 -157 aa; GenBank EMQ93586.1 LysM domain protein [S. pneumoniae]), and PsaA (238-309 amino acids (aa); GenBank AAF70667.1 PsaA [S. pneumoniae]) was previously described [35]. The purified modified protein expressed from this plasmid was used as the control bacterial antigen in this study [36, 37].
A rabbit hyperimmune sera to the recombinant PspA-Spr1895-PsaA protein was generated as follows. Three female rabbits aged 3-4 months and weighing 2 to 2.5 kg were sourced from the laboratory breeding nursery of the Russian Academy of Sciences in Rappolovo, Leningrad Region, Russia. They were maintained under standard conditions: 0.5 m2 cage space, 0.4 m height, 15-21°C, 45-65% humidity, 12-hour light/dark cycle, solid/slatted floors with bedding, and enrichment items. The rabbits were allowed ten days to be acclimated to the housing facility. All animals were kept in a specialized pathogen-free facility, receiving autoclaved food and water ad libitum. For the immunization process, recombinant PspA-Spr1895-PsaA protein was dissolved in phosphate-buffered saline (PBS) and mixed with Adjuvant Aluminum (Sigma Aldrich, St. Louis, MO, USA) in a 2:1 ratio. The protein was subcutaneously administered to rabbits at a dose of 100 μg in a total volume of 200 μl on days 1, 21, and 35 from the beginning of the experiment. Blood samples were collected on the 42nd day from the ear vein, and sera were separated by centrifugation of the blood at 3500 rpm for 15 min.
Clinical isolate No.73 of S. pneumoniae, serotype 3, was grown at 37°C for 18 h in THB medium supplemented with 20% horse serum (Thermo Fisher Scientific Waltham, MA, USA) under anaerobic conditions. Moreover, to count the number of bacteria, a dense medium consisting of Columbia agar, 5% defibrinated sheep blood, and 10% horse serum was used. Bacterial titers were expressed as a number of colony-forming units (CFU) per mL.
2.2. Design of Modified HA+PspA Gene
The gene encoding the H7N9 HA protein was modified by inserting the bacterial PspA gene (160-262 аa) between the HA1 subunit and the signal peptide (SP) via flexible linkers (AAAPGAA). The modified HA was constructed through a series of PCR amplifications using primers listed in Table 1. Briefly, a bacterial fragment was amplified in two steps to introduce flexible linkers AAAPGAA from both N- and C-ends, as well as sites for the BsmBI restriction enzyme. In parallel, BsmBI restriction sites were introduced from the C-terminus of the signal peptide and the N-terminus of the HA1 subunit using two PCR reactions with specially designed primers (Table 1). Then, these two fragments were combined in a common PCR with forward and reverse primers to obtain the full-length HA+BsmBI gene. The condition for the polymerase chain reaction were as follows: 1)Initial DNA denaturation 98°C (30s) → cycle entry 2)DNA denaturation 98°C (10s) → 3) primer annealing 55°C (30s) → 4) elongation of single-stranded DNA 72°C (30sec/1kb) → end of cycle, back to step 2 (35 cycles) → 5)final elongation 72°C (10min) → 4°C (storage). PCR was performed using the Phusion Green High-Fidelity DNA Polymerase kit (Thermo Scientific) according to the manufacturer's instructions. All amplicons that matched the size of the PCR products were extracted from the agarose gel using a kit for extraction and purification of DNA (Cleanup Mini, Eurogen, Russia).
Treatment of the modified bacterial fragment and the HA+BsmBI gene with BsmBI restriction enzyme, followed by ligation with T4 ligase, resulted in the HA+PspA modified gene encoding the bacterial fragment from the N-terminus of the HA1 subunit. Finally, the HA+PspA modified gene was amplified with primers containing additional sites for SapI restriction enzyme and was then cloned into the vector for influenza virus reverse genetics (pCIPolISapIT, CDC, Atlanta, USA) by treating with SapI enzyme and ligating with T4 DNA ligase.
2.3. Generation of the Recombinant Influenza Virus
The rescue of the recombinant LAIV/HA+PspA virus was performed by transfecting Vero cells with the required set of plasmid DNAs encoding PB2, PB1, PA, NP, M, and NS genes of Len/17 MDV, the NA gene from the A/Anhui/1/2013 (H7N9) strain, and the modified HA+PspA gene. The transfection was performed by electroporation of Vero cells using the Neon Transfection System (Thermo Fisher Scientific, Waltham, MA, USA) in the mode of two pulses of 1150V for 20 ms, according to the instructions of the manufacturer. Three days post-transfection, the resuspended cells, and media were inoculated into 10-day-old chicken embryos, and the viable virus was harvested after 48 hours of incubation at 33°C. The presence of the virus was determined by a hemagglutination assay using 0.5% of chicken red blood cells. Working virus stock was prepared by an additional passage in eggs at 33°C (E2). The allantoic fluid was harvested, clarified by low-speed centrifugation, and stored in single-use aliquots at -70°C.
| Amplicon | Forward | Reverse | Product Size bp |
|---|---|---|---|
| Addition of flexible linker AAAPGAA and BsmBI restriction site to the bacterial PspA fragment | |||
| PspA+linker (1 step) | GCAGCACCTGGTGCAGCAGCTGACCTGAAAAAAGC | TGCACCAGGTGCTGCTGCCCACATACCGTTTTCC | 349 |
| linker+BsmBI site (2 step) | TATACGTCTCAGCAGCAGCACCTGGTGCA | TATACGTCTCTTGCTGCACCAGGTGCTGCTGC | 377 |
| Addition of BsmBI restriction site between SP and HA1 subunit of HA gene | |||
| HA1/2+BsmBI | GAGACGGTACGTCTCCAGCAGACAAAATCTGCCTCG | ACTGGCTCTTCTATTAGTAGAAACAAGGGTGTTTT | 1694 |
| SP+BsmBI | GATCGCTCTTCAGGGAGCAAAAGCAGGGG | GAGACGTACCGTCTCGCTGCTGCATTTGTTGGAATG | 111 |
| SP+BsmBI+HA1/2 | GATCGCTCTTCAGGGAGCAAAAGCAGGGG | ACTGGCTCTTCTATTAGTAGAAACAAGGGTGTTTT | 1789 |
| Addition of SapI restriction sites to both ends of the modified HA+PspA gene | |||
| HA+PspA+SapI | GATCGCTCTTCAGGGAGCAAAAGCAGGGG | ACTGGCTCTTCTATTAGTAGAAACAAGGGTGTTTT | 2118 |
2.4. In vitro Studies
2.4.1. Growth Characteristics in Eggs, Ts/Ca Phenotypes
Moreover, to determine infectious viral titers in eggs, chicken embryos were inoculated with 10-fold dilutions of the virus. The eggs were incubated at various temperatures: at 33°C and 38°C for 3 days and at 26°C for 6 days. The presence of the virus was determined by incubation of allantoic fluid with 0.5% chicken red blood cells, and the titer was calculated by the Reed and Muench method and expressed in log10EID50/mL [13]. The temperature sensitive (ts) phenotype was analyzed by comparing viral titers at 33°C and 38°C, whereas the cold-adapted (ca) phenotype was determined by comparing viral titers at 33°C and 26°C.
2.4.2. Genetic Stability
The genetic stability of the recombinant vaccine strain LAIV/HA+PspA was studied after 10 sequential passages of eggs incubated at 33°C. After each passage, an analytical PCR analysis was performed to confirm the presence of the PspA coding region within the HA gene; at the last passage, whole genome Sanger sequencing was performed to confirm the absence of unwanted mutations within the insert and the presence of a full set of attenuating mutations of Len/17 MDV. Sequencing was run on the ABI 3130xl genetic analyzer (Applied Biosystems Japan Ltd., Tokyo, Japan) using the BigDye v3.1 cycle sequencing kit (Life Technologies, Austin, TX, USA).
2.4.3. Replication on MDCK Cells
Ten-fold dilutions of the virus were prepared on DMEM medium supplemented with 1× antibiotic-antimycotic (Capricorn, Germany) and 1 μg/mL of TPCK-treated trypsin (Sigma-Aldrich, St. Louis, MO, USA). MDCK monolayers in a format of 96-well plates were inoculated with virus dilutions, 25 μl per well, six wells per dilution. After adsorption for 1 hour, the inoculum was removed, followed by the addition of 150 μl of the same culture medium. The infected cells were incubated at 33°C, 5% CO2, for 3 days. Then, 50 μl of the medium was taken from each well and mixed with 50 μl of 0.5% chicken red blood cells to detect the virus. Viral titers were determined by the Reed and Muench method [38] and expressed in log10TCID50/mL.
For generation of growth curves, MDCK cells grown on 6-well plates were inoculated with the virus at a multiplicity of infection 0.01 using the same procedure as described above. The cells were incubated at 33°C, and the samples were collected every 12 hours and stored at -70°C for subsequent titration by the TCID50 assay as described above.
2.4.4. Expression of Bacterial Protein PspA
Expression of correctly folded bacterial PspA fragment as a part of modified HA protein of influenza virus was confirmed by Western Blot analysis of sucrose gradient purified LAIV/HA+PspA virus, in comparison with purified classical H7N9 LAIV strain. The separation of viral proteins was carried out by electrophoresis under denaturing conditions. A ready-to-use 12% Mini-PROTEAN® TGX ™ Precast Protein Gels (Bio-Rad, Hercules, CA, USA) were used for polyacrylamide gel electrophoresis (PAGE). The gels were run at 100 V for 1 hour, and one gel was further stained with Coomassie (QC Colloidal Coomassie Stain) (Bio-Rad, CA, USA). Another gel was used for protein transfer to a nitrocellulose membrane using semi-dry electroblotting at 90-100 mA for 1 hour, followed by membrane incubation with primary antibodies diluted in blocking buffer (3% milk in PBS with 0.05% Tween 20), specific either to influenza virus subtype A/Anhui/1/2013 (H7N9) (mouse serum) or to S. pneumonia (hyperimmune rabbit sera to the pneumo- coccal hybrid protein PspA-Spr1895-PsaA). After incubation and washing, secondary species-specific antibodies conjugated with horseradish peroxidase were added, followed by additional incubation for 1 hour. The membranes were then developed with a TMB Substrate Kit (Vector Laboratories, Burlingame, CA, USA), washed with tap water, and air dried.
2.5. Mouse Study Design
Six to eight-week-old old female BALB/c mice were purchased from the nursery of the Russian Academy of Sciences “Stolbovaya” (Moscow region, Russia). Before starting the experiments, the animals were quarantined for at least 2 weeks. The animals were kept on a standard diet and had free access to water and food.
Mice were immunized intranasally with two doses of either recombinant LAIV/HA+PspA virus or control LAIV H7N9 virus, three weeks apart. Vaccine viruses were inoculated at a dose of 106 EID50 in a volume of 50 µl. Mock-immunized animals received PBS. On day 3 of the experiment, replication of the vaccine viruses in nasal turbinates and lungs (n=4) was assessed by titration of tissue homogenates in eggs. The serum samples were collected from a group of mice (n=6) for immunogenicity studies. Four weeks after the second immunization, mice were challenged with Sh/PR8 (H7N9) or X-79 (H3N2) virus at a dose of 1×105 EID50, in a volume of 50 µl, and on day 3 post-challenge, the mice were infected with S. pneumoniae at a dose of 6.0×104 CFU, thus modelling the bacterial complications of the influenza infection. The protective efficacy was evaluated by monitoring body weight and survival rates during two weeks post influenza infection (Fig. 1).
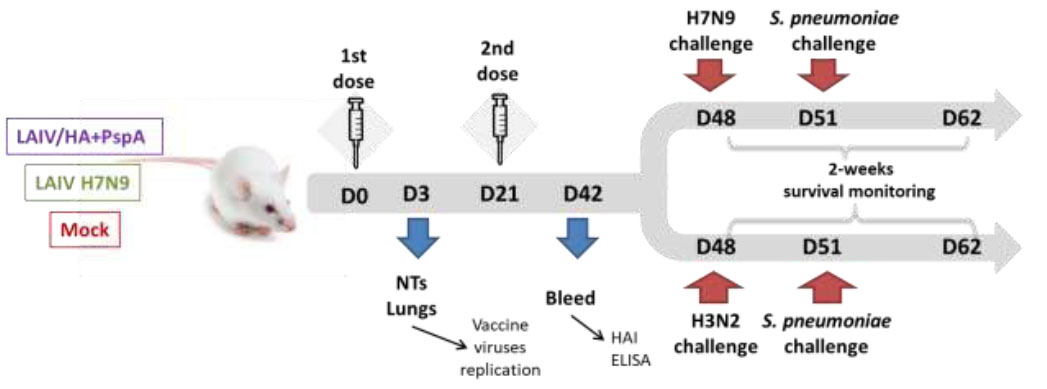
Mouse study design. Groups of 20 mice were immunized with either bivalent experimental vaccine LAIV/HA+PspA or control LAIV H7N9 virus; control mice received PBS. NTs – nasal terminates.
2.6. Immunogenicity Studies
The serum antibody responses were measured by hemagglutination inhibition assay (HAI), and enzyme-linked immunosorbent assay (ELISA) in samples was collected three weeks after the second vaccine dose.
2.6.1. ELISA
ELISA was performed against two antigens – (i) LAIV H7N9 sucrose gradient purified whole virus and (ii) a hybrid pneumococcal recombinant protein (PspA-Spr1985-PsaA), using a standard protocol. Briefly, high-binding 96-well microplates (Corning, NY, USA) were coated with 16 hemagglutinating units of influenza virus antigen or with 100 ng of the pneumococcal protein. After blocking and washing, two-fold dilutions of the mouse sera were added to the plates, followed by incubation for 1 h at 37°C, washing, and the addition of goat anti-mouse IgG secondary antibody conjugated with horseradish peroxidase (Bio-Rad, CA, USA). After additional incubation and washing, the plates were developed using a 1-step TMB ELISA solution (Thermo, Rockford, IL, USA). The results were read using an xMark Microplate reader (BioRad, CA, USA) at 450 nm. Antibody titers were defined as the maximum serum dilution with an OD450 value exceeding the control values (no serum added) at least two times.
2.6.2. Hemagglutination Inhibition Assay
For the HAI assay, serum samples were treated with a receptor-destroying enzyme (Denka Seiken, Japan) to remove nonspecific inhibitors, according to the instructions of the manufacturer. The HAI reaction was performed in U-bottom 96-well plates using the H7N9 LAIV virus as antigen, according to the method described in a study [39]. Mouse sera were serially diluted in PBS, and 4 HA units of the virus were added to each well and incubated for 1 hour at room temperature. Then, an equal volume of 0.5% chicken red blood cells was added to visualize the presence of the virus. HAI titer was defined as the last serum dilution where complete inhibition of hemagglutination occurred.
2.7. Statistical Analysis
Statistical analysis was performed using GraphPad Prism software (version 6.0). Two groups of values were compared using the Mann-Whitney U-test. For comparisons of three independent groups, one-way ANOVA with Dunnett's multiple comparisons test was used. Survival rates between different study groups were compared with the logrank Mantel-Cox test. Differences with p < 0.05 were considered significant.
3. RESULTS
3.1. Generation and an in vitro Characterization of the Recombinant Influenza Virus Expressing PspA Fragment
A pneumococcal vaccine candidate based on live influenza vaccine was rescued by standard gene engineering methods. PspA gene fragment flanked by flexible linkers was inserted into the HA influenza gene, resulting in the exposure of the S. pneumoniae antigen on the surface of the influenza virus as part of the modified hemagglutinin (Fig. 2A-E). The amplification of the HA fragment demonstrated that the bacterial PspA fragment gene was successfully inserted into the influenza HA gene (Fig. 2C). The LAIV/HA+PspA recombinant virus was rescued in Vero cells with amplification in eggs. The virus population was further cloned by limiting dilutions in eggs to ensure the purity of the viral stock. The presence of the PspA fragment insert within the modified HA gene was confirmed by Sanger sequencing the full-length HA gene. Furthermore, western blot analysis of the purified recombinant virus, along with the control classical LAIV strain, using polyclonal antibodies to pneumococcal hybrid protein revealed specific bands with molecular mass ~60 and ~90 kDa, corresponding to the HA0 precursor of the HA and HA1 subunit only in the recombinant LAIV/HA+ PspA virus, but not in control H7N9 LAIV strain (Fig. 2D). These results indicate the co-expression of the bacterial polypeptide within HA in the virion.
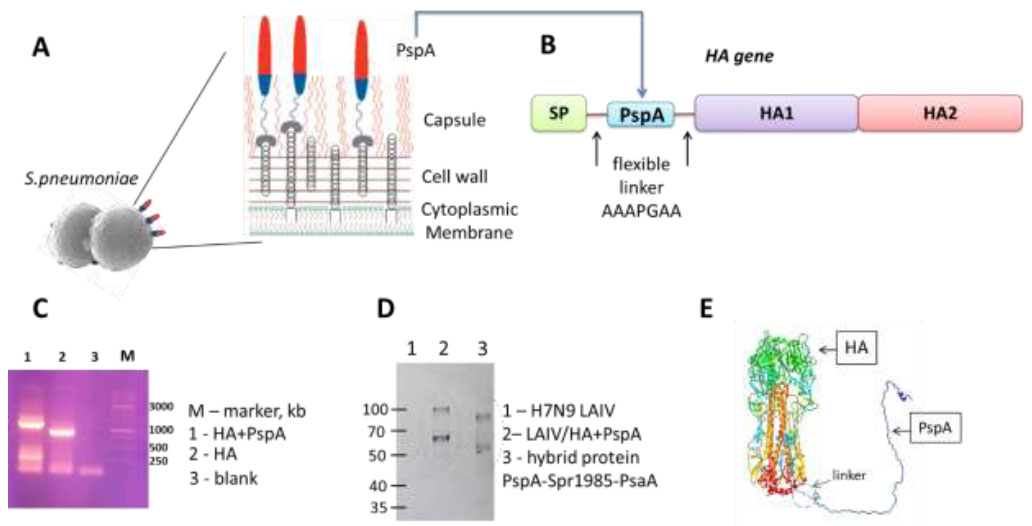
Overall design of the bivalent vaccine construct and characterization of the modified HA+PspA gene and protein. (A) Schematic representation of PspA on the surface of S. pneumoniae. Red and blue indicate the polar charges of PspA: negatively charged, distant from the surface, and positive, respectively. (B) Scheme of the modified HA hemagglutinin protein with the bacterial PspA insertion. (C) Electrophoresis with amplifying HA fragment gene; 1-HA+PspA, 2-HA, 3-blank, M-marker (DNA Ladder 1 kb, Eurogen, Russia) (D) Immunoblotting with polyclonal antibodies to pneumococcal hybrid protein PspA-Spr1985-PsaA; M-marker (kDa), 1-H7N9 LAIV, 2-LAIV/HA+PspA, 3-hybrid protein PspA-Spr1985-PsaA. (E) 3D model of the hemagglutinin molecule with the bacterial PspA insertion (one monomer shown). Linker – AAAPGAA, SP - signal peptide.
Ten sequential passages of the recombinant LAIV/HA+PspA strain revealed high genetic stability of the construct since no mutations were detected in the modified HA+PspA gene in the passaged variant. In addition, all attenuating mutations located in the genes derived from the Len/17 master donor virus were preserved (data not shown).
The recombinant LAIV/HA+PspA virus grew in eggs at 33°C at a titer of 8.5 lgEID50/mL, suggesting that such a recombinant vaccine can be successfully manufactured using this substrate (Table 2). Assessment of virus replication at different temperatures revealed that the LAIV/HA+PspA virus is temperature sensitive, i.e., incapable of active replication at elevated temperatures (38°C), similar to the control LAIV H7N9 strain (Table 2). Although the recombinant vaccine strain carrying a bacterial insertion had a slightly reduced titer at all incubation temperatures compared to the classical LAIV H7N9 strain, it was characterized by both ts and ca phenotypes that are characteristics of Len/17-based live attenuated vaccine reassortant viruses (Table 2).
| Virus | Virus Titer at Indicate Temperatures, lg EID50/mL | Phenotype | ||
|---|---|---|---|---|
| 26°С | 33°С | 38°С | ||
| H7N9 LAIV | 6.2 | 9.3 | 3.3 | ts, ca |
| Len/17 | 7.0 | 9.0 | 3.2 | ts, ca |
| LAIV/HA+PspA | 5.1 | 8.5 | 2.0 | ts, ca |
Growth kinetic of recombinant vaccine LAIV/HA+PspA and classical LAIV H7N9 virus on MDCK cells at a multiplicity of infection 0.01. The graph shows the mean and standard deviation of three values.
Comparison of the growth curves of the LAIV/HA+ PspA recombinant virus and the H7N9 LAIV control strain on MDCK cells revealed the impairment of virus replicative potential when the PspA fragment was inserted into HA gene (Fig. 3). Probably, the expression of the modified HA+PspA gene by the recombinant virus results in slower viral replication or viral particle assembling, thus leading to slower viral accumulation in the culture medium. Probably, several adaptive passages on this cell line might improve viral fitness and increase virus infectivity in this substrate.
3.2. Replication of LAIV/HA+PspA Recombinant Virus in Mouse Respiratory Tract
Groups of BALB/c mice were intranasally inoculated with 106 EID50 of the recombinant vaccine LAIV/HA+PspA or the classical LAIV H7N9. On day 3, the intensity of viral replication in the upper (nasal turbinates) and the lower (lungs) respiratory tract was determined as a measure of vaccine virus infectivity and attenuation. As expected, both LAIV/HA+PspA and H7N9 LAIV viruses replicated well in NTs, but no replication was detected in the lungs (Fig. 4). These data confirmed that the LAIV/HA+PspA recombinant vaccine prototype retained the attenuated phenotype-specific to the LAIV strains, and the infectivity of the virus for the mouse upper respiratory tract was not compromised by the insertion of the bacterial PspA fragment into the LAIV genome.
3.3. Immunogenicity of the Recombinant LAIV/HA+PspA
It is necessary to establish whether the presence of a foreign insertion in the vaccine virus influences the formation of an immune response to the influenza virus, as well as whether this insertion is capable of inducing immunity against Streptococcus pneumoniae. Three weeks after the second immunization, serum samples were collected from a subset of mice (n=6) to determine serum antibody titers to influenza virus in HAI assay and ELISA, as well as to the pneumococcal hybrid protein in ELISA. As shown in Fig. (5A, B), both the recombinant virus and the control LAIV strain induced similar levels of influenza-specific antibody responses. Although the mean levels of HAI and IgG antibodies were slightly lower in the recombinant vaccine group compared to the LAIV H7N9 group, these differences were not statistically significant. In contrast, only the recombinant vaccine LAIV/HA+PspA was able to induce detectable levels of IgG antibodies to the antigen of S. pneumoniae (Fig. 5C), while no antibodies were found after immunization with classical LAIV virus. Therefore, intranasal administration of the recombinant LAIV/HA+PspA vaccine prototype stimulated the production of serum IgG antibodies both to influenza virus and bacterial antigens.
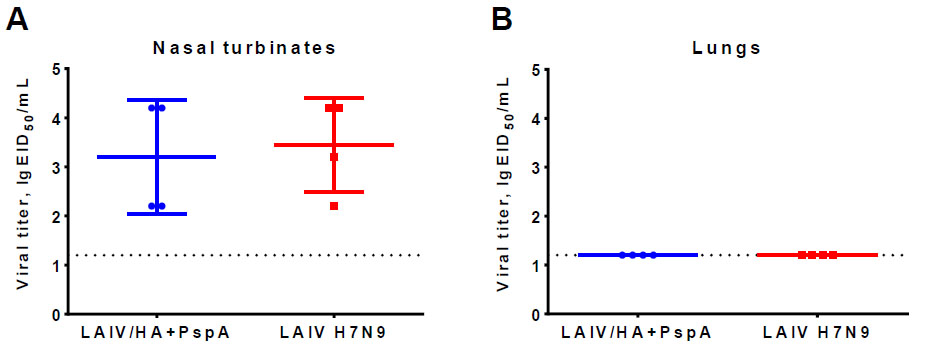
Infectious titer of recombinant LAIV/HA+PspA candidate and classical LAIV H7N9 vaccine virus in nasal turbinates (A) and in the lungs (B) of mice on day 3 after intranasal immunization at a dose of 106 EID50.
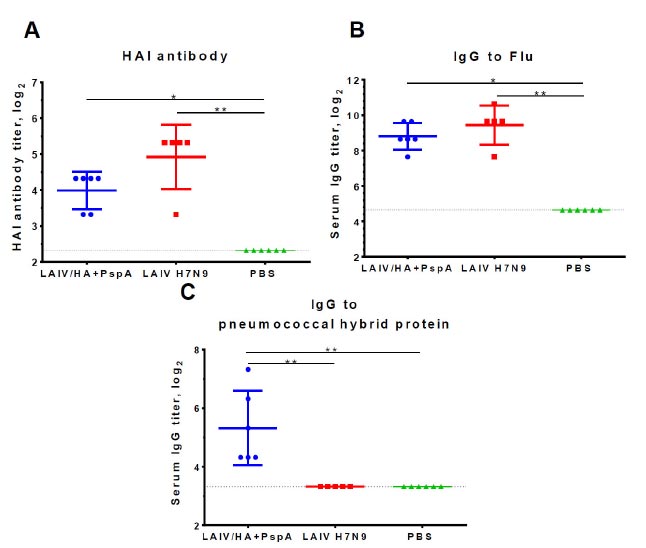
Serum antibody responses to influenza virus measured in HAI assay (A) or ELISA (B), as well as to the pneumococcal hybrid protein PspA-Spr1895-PsaA in ELISA (C). The mice were intranasally immunized twice with 106 EID50 of the indicated vaccine virus or with placebo (PBS) at a 3-week interval. Blood samples were collected three weeks after the second dose. The data were analyzed using the Kruskal-Wallis test with Dunn’s correction for multiple comparisons. *p<0.05; **p<0.01.
3.4. Protective Activity of the LAIV/HA+PspA Recombinant Vaccine Prototype
Influenza-specific immune responses induced by the two-dose immunization with the recombinant vaccine were sufficient for complete protection of mice against lethal influenza challenge infection caused by a homologous influenza virus, whereas 100% of mock-immunized mice succumbed to the infection by day 9 post-challenge (Fig. 6). These data suggest that the insertion of the bacterial antigen into LAIV genome did not affect the anti-influenza protective potential of the vaccine. Importantly, when immunized mice were subjected to homologous or heterologous influenza challenge with subsequent S. pneumonia co-infection (bacterial infection occurred on day 3 after influenza challenge), significant morbidity was observed in all study groups, with 100% lethality detected in PBS and H7N9 LAIV groups and 40% survival rates were observed in the LAIV/HA+PspA bivalent vaccine prototype (Fig. 7). These findings indicate that PspA-specific antibody responses induced by this vaccine candidate could have contributed to partial protection of immunized mice against influenza infection with severe bacterial coinfection. However, further studies are needed to estimate the levels of PspA-specific antibodies that are required for such protection.
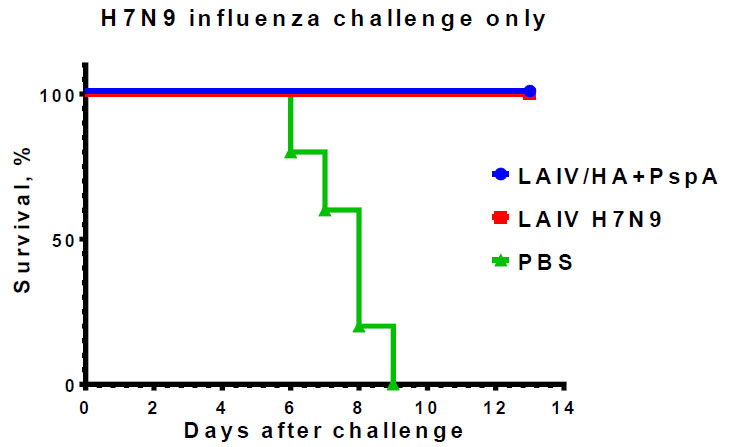
Protective potential of LAIV/HA+PspA and classical LAIV H7N9 against homologous challenge virus Sh/PR8 (H7N9). Mice (n=5) were immunized with two doses of the indicated virus or PBS and challenged on day 48 of the study. The survival rates were monitored for 13 days post-challenge.
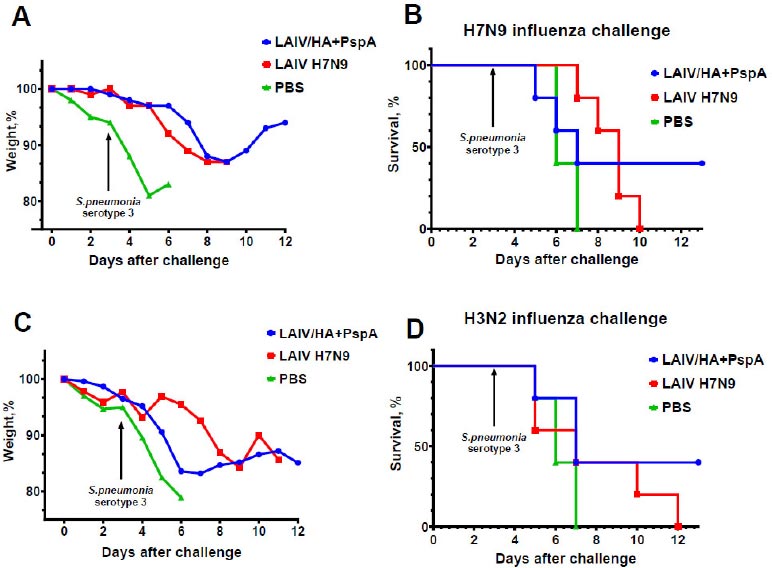
Protective potential of LAIV/HA+PspA and classical LAIV H7N9 against influenza infection complicated by bacterial co-infection. The mice (n=5) were immunized with two doses of indicated virus or PBS and challenged with influenza virus Sh/PR8 (H7N9) (A, B) or X-79 (H3N2) (C, D). On day 3 after infection, the same mice were infected with S. pneumoniae serotype 3 (strain 73). Weight loss and survival rates were monitored for 12 days post-influenza challenge.
4. DISCUSSION
Secondary bacterial infection with S. pneumoniae after influenza virus infection increases the severity of illness and leads to life-threatening pneumonia. Infection with the influenza virus causes damage to mucosal epithelial cells and exposure of receptors, which facilitates bacterial adhesion and dysfunction of immune effectors, allowing bacteria to enter the lower respiratory tract [40]. Treatment with antibiotics for secondary bacterial pneumonia is not always successful. Damage to the wall of the gram-positive bacterium S. pneumoniae by the recommended β-lactam antibiotics leads to the release of pro-inflammatory bacterial components, exacerbating the course of the infection [31]. It is thought that the adminis- tration of the influenza vaccine alone can prevent secondary pneumococcal infection because by avoiding infection with the influenza virus, the secondary bacterial invasion will be minimized [32]. However, the high variability of the influenza virus requires an annual reformulation of the influenza vaccine composition and does not prevent the emergence of antigenic influenza virus strains different from those included in the vaccine. Therefore, the inclusion of a bacterial antigen in the influenza vaccine would reduce the severity of compli- cations and mortality caused by secondary bacterial infection.
In contrast to the immune responses raised to pneumococcal capsular polysaccharide antigens, the responses to the pneumococcal surface protein A are highly cross-reactive and can react with different pneumo- coccal serotypes. For example, serum from a rabbit immunized with native PspA protein was shown to recognize 95 different pneumococcal isolates containing 16 capsular serotypes [41]. Therefore, the PspA protein is considered a promising target for the development of next-generation pneumococcal vaccines. Indeed, immunization of humans with the recombinant PspA protein induced broadly cross-reactive antibodies to heterologous PspA molecules that passively protected mice against a lethal dose of S. pneumoniae [42, 43]. However, the immuno- genicity of PspA delivered by a viral vector may be limited by the immunogenicity of the vector or by pre-existing immunity to the vector [44].
In the current study, we used live attenuated influenza vaccine virus as a vector to deliver PspA fragments to the target cells to elicit antibody responses to this bacterial surface protein. In the case of using the influenza virus as a vector, the vector-specific immunity will be considered as an advantage since influenza viruses circulate globally and cause annual epidemics. Importantly, in this case, it is easy to manipulate the vector antigenic properties by simply replacing the HA and NA proteins with a more recent influenza virus, thus avoiding pre-existing antiviral immunity [45]. In our development, the foreign antigen was inserted into the open reading frame of the influenza HA molecule in such a way that the PspA protein appeared on the surface of the viral particle as a part of the HA+PspA-modified protein. The correct folding of the inserted bacterial antigen was confirmed by Western blot analysis with polyclonal rabbit antibodies raised to a recombinant hybrid protein consisting of three S. pneumoniae polyproteins (PspA, Spr1895 and PsaA). This modified LAIV strain retained all major properties of the attenuated influenza vaccine virus, such as temperature-sensitive and cold-adapted phenotypes, as well as attenuation for mice. Importantly, the LAIV/HA+PspA recombinant vaccine strain was genetically stable after 10 passages in eggs, meaning that the insert and all required attenuation mutations were present in the passaged variant.
Two-dose intranasal immunization of mice with the recombinant LAIV/HA+PspA virus-induced influenza-specific antibody responses similar to that of classical LAIV strain; in addition, detectable levels of PspA-specific antibodies were found in serum samples of vaccinated mice. Although these bacterial protein-specific antibody levels were insufficient to fully protect mice against lethality caused by influenza-bacterial coinfection, a 40% survival rate was achieved in the LAIV/HA+PspA group, compared to 100% lethality in the group immunized with standard LAIV.
In a study by Katsura et al., replication-incompetent influenza viruses carrying the PspA antigenic region instead of HA were engineered [46]. These viruses are replicative incompetent due to the lack of HA protein but can produce viral proteins once entering the cells; therefore, the PspA target antigen was also produced along with viral proteins, thus eliciting specific antibodies. This vaccine fully protected mice against lethal infection with S. pneumoniae, compared to the control groups, by significant reduction of bacterial carriage in the upper respiratory tract [46]. This study demonstrated the feasibility of creating such bivalent vaccines based on influenza viruses; however, such vaccines will have limited protective capacity against influenza virus infection since no anti-HA antibodies are induced. Accordingly, low protection is expected against viral-bacterial super- infections.
Partial non-specific protection by influenza vaccines against Streptococcus bacteria is well known. Immuni- zation with live and inactivated influenza vaccines induces neutralizing antibodies, IgG antibodies of various isotypes, as well as IgAs on mucosal tissues. Immunization with influenza vaccines limits the production of pro-inflam- matory cytokines (IL-6, TNF-α, IFN-γ), the synthesis of which is induced by secondary infection with Streptococci following influenza infection. Inactivated, as well as live influenza vaccines, limit morbidity and mortality following secondary infection with Streptococcus (S. pyogenes) following influenza. A study [47] showed that BALB/c mice immunized with influenza vaccines had 67 and 78% survival (IIV, LAIV, respectively) to superinfection (influenza + bacteria), whereas naive mice had only 18% survival. Similarly, no significant weight loss during superinfection was observed in the vaccine groups. In another recent study, a two-dose immunization with trivalent LAIV was shown to partially protect BALB/c mice against sequential infection with S. pneumoniae following the influenza virus, reaching a survival rate of 80% [48]. The level of bacterial load in the lungs, as well as the number of immunized mice with confirmed bacteremia are significantly lower than that of unvaccinated animals. All these findings confirm that superinfection can be limited by immunity induced by influenza virus vaccine, although prevention of disease is not complete. However, protection against bacterial infection by influenza vaccination alone is not possible because of the lack of definite immunity against bacteria. Therefore, the induction of antibodies against S. pneumoniae is important to prevent such bacterial infections.
Besides PspA, other conserved pneumococcal proteins can be considered for vaccine design. In fact, 26 out of 65 protein virulence factors of pneumococcus are expressed constitutively and have no significant variability among different strains [49], seven of which are used to develop protein-based vaccines against pneumococcal disease (among them PsaA, pneumolysin, and serine-threonine kinase protein, StkP) [50]. The insufficient immunogenicity of PspA within the influenza vector can be overcome by combining more than one protein antigen into a single viral particle. For example, PsaA and PspA proteins can be inserted into the LAIV virus genome, as it was shown previously that intranasal immunization with a mixture of PsaA and PspA could provide significant protection against nasopharyngeal carriage of S. pneumoniae [51, 52].
CONCLUSION
The presented design of a recombinant influenza virus carrying immunogenic fragments of a bacterial pathogen can be considered a promising strategy for the combined protection of vaccinated individuals against influenza and its bacterial complications, and further in-depth studies of such recombinant viruses in preclinical studies are warranted.
AUTHORS’ CONTRIBUTION
It is hereby acknowledged that all authors have accepted responsibility for the manuscript's content and consented to its submission. They have meticulously reviewed all results and unanimously approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| LAIV | = Live Attenuated Influenza Vaccine |
| MDV | = Master Donor Virus |
| Ts | = Temperature Sensitive |
| Ca | = Cold adapted |
| EID50 | = 50% chicken embryo infection dose |
| TCID50 | = 50% tissue culture infection dose |
| HAI | = Hemagglutinin Inhibition |
| NTs | = Nasal Turbinates |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The animal study protocol was approved by the Ethics Committee of the Institutional Local Ethical Committee (IEM, Saint Petersburg, Russia) (1/21 was approved on 28.01.21).
HUMAN AND ANIMAL RIGHTS
All of the experiments on mice, rabbits, and chicken embryos were performed according to European Union legislation. At the end of the study, the animals were humanely euthanized.
This study adheres to internationally accepted standards for animal research, following the 3Rs principle. The ARRIVE guidelines were employed for reporting experiments involving live animals, promoting ethical research practices.
AVAILABILITY OF DATA AND MATERIALS
The data presented in this study are available on request from the corresponding author [A.M].
FUNDING
This research was funded by the Russian Ministry of Science and Higher Education under the project FGWG-2022-0001.

