All published articles of this journal are available on ScienceDirect.

Heterologous Expression of Histidine Acid Phytase From Pantoea sp. 3.5.1 in Methylotrophic Yeast Pichia Pastoris
Abstract
Background and Objective:
The major storage form of phosphorus in plant-derived feed is presented by phytates and not digested by animals. Phytases are able to hydrolyze phytates and successfully used as feed additives. Nevertheless, nowadays, there is a constant search of new phytases and expression systems for better production of these enzymes. In this study, we describe cloning and expression of gene encoding histidine acid phytase from Pantoea sp. 3.5.1 using methylotrophic yeast Pichia pastoris as the host.
Methods:
The phytase gene was placed under the control of the methanol-inducible AOX1 promoter and expressed in P. pastoris. Experiments of small-scale phytase expression and activity assays were used to test recombinant colonies. Four different signal peptides were screened for better secretion of phytase by P. pastoris. After 36 h of methanol induction in shake flasks, the maximum extracellular phytase activity (3.2 U/ml) was observed in P. pastoris strain with integrated construct based on pPINK-HC vector and Kluyveromyces maxianus inulinase gene signal sequence. This phytase was isolated and purified using affinity chromatography.
Results:
Recombinant phytase was a glycosylated protein, had a molecular weight of around 90 kDa and showed maximum activity at pH 4.0 and at 50°C. Recombinant phytase had excellent thermal stability – it retained high residual activity (100% ± 2%) after 1 hour of heat treatment at 70°C.
Conclusion:
The enhanced thermostability of the recombinant phytase, its expression provided by strong inducible promotor and the effectively designed expression cassette, the simple purification procedure of the secreted enzyme, and the possibility of large-scale expression make the foundation for further production of this bacterial phytase in P. pastoris at an industrial scale.
1. INTRODUCTION
According to the US Department of Agriculture, the most produced type of meat in 2019 was poultry - 125.6 million metric tons. The fast-growing poultry industry requires a huge amount of plant-derived feeds [1]. A significant portion of phosphorus in feed grains is present as phytic acid and its insoluble complexes with metal ions – phytates. Such complexes cannot be utilized by animals, leading to the problem of insufficient quantity of available phosphorus in animal feeds. Similar problems with phosphorus availability are common for all monogastric animals; thus, increased amounts of inorganic phosphate are used in feed supplements. The unutilized feed phytate is largely excreted with animal manure and can cause environmental pollution and eutrophication of water reservoirs [2].
Microbial enzymes – phytases - are able to hydrolyze insoluble phytates with the release of free phosphates together with the chelated metal ions such as calcium, iron, copper and others and are ubiquitous enzymes in the animal nutrition industry [3]. Initially, the commercial production of phytases was concentrated mainly on fungi phytases. However, bacterial phytases are more promising due to their better catalytic efficiency, broad substrate specificity and greater resistance to proteolysis [4, 5].
All phytases are divided into two groups - acidic and alkaline enzymes, depending on their pH-profile. Acidic phytases cover 4 classes of enzymes - histidine acid phytases (HAPs), purple acid phytases (PAPs, typical for plants) and recently identified protein tyrosine phosphatases (PTP-like inositol polyphosphatases). Alkaline phytases are represented by extracellular beta-propeller phytases (BPPs) of gram-positive spore-forming bacteria of the Bacillus genus [5]. Nowadays, all phytase-based feed additives on the market are produced using histidine acid phytases, which possess high phytate-hydrolyzing activity in the conditions of monogastric animal’s stomach (specific pH and temperature) [6]. Phytases belonging to HAPs family share similar features such as pH-optimum at acidic pH (2.5-6.5), temperature optimum around 50-60 °С and great proteolytic resistance [1]. To date, there are two major phytases used in animal nutrition - the phytases from Aspergillus niger and Escherichia coli (AppA). Still, the thermal stability of E. coli AppA phytase is insufficient to go through the feed pelleting process [7]. Feed additives are produced at extreme temperatures (85-90 ° C); therefore, the thermal stability of phytases is one of the crucial features in their manufacturing [8]. Moreover, ‘ideal’ phytases for animal nutrition must meet several requirements. They should be effective in phytate degradation in the gastrointestinal tract, stabile and active under the pH of the digestive tract, resistant to proteolysis, stable to high temperatures while feed processing, resist inactivation by long-term storage and cheap to produce [3]. Certainly, the properties of recombinant enzymes depend on the expression system used. Alteration of phytases resulting in the increased thermal or pH stability, will be beneficial for their application as animal feed additives [9].
Methylotrophic yeast Pichia pastoris is a handy tool for heterologous gene expression, producing high level of recombinant proteins and is widely used for large-scale production of different enzymes for various applications [10]. P. pastoris is capable of growing on simple media and achieves high cell density. This is due to the fact that P. pastoris is obligate aerobe and belongs to the so-called “Crabtree negative” types of yeast. They are characterized by respiratory growth type, which distinguishes P. pastoris from the related Saccharomyces cerevisiae yeast, since the latter utilize glucose mainly due to glycolysis and produce ethanol and many by-products, which negatively affect the growth of culture [11]. This expression system allows effective control of the transcription level of target genes using strong and tightly regulated inducible promoters [12]. Moreover, being a eukaryotic expression system, P. pastoris is able to conduct processing, folding and posttranslational modifications of heterologous proteins [13]. The advantage of P. pastoris is the nature of N-glycosylation of secreted proteins. Unlike S. cerevisiae, P. pastoris is not prone to hyperglycosylation, which is often accompanied by changes in protein properties, makes it extremely immunogenic, and limits the use of recombinant proteins in the pharmaceutical and food industries [14]. In addition, cloning of the genes of interest by homologous recombination between the vector and yeast genome allows us to obtain very stable transformants [15]. Various phytases have successfully been expressed and produced using P. pastoris system [16-18].
In this paper, we report the molecular cloning of the histidine acid phytase agpP gene from Pantoea sp. 3.5.1 and the expression of the corresponding protein in P. pastoris.
2. MATERIALS AND METHODS
2.1. Strains, Plasmids and Medium Composition
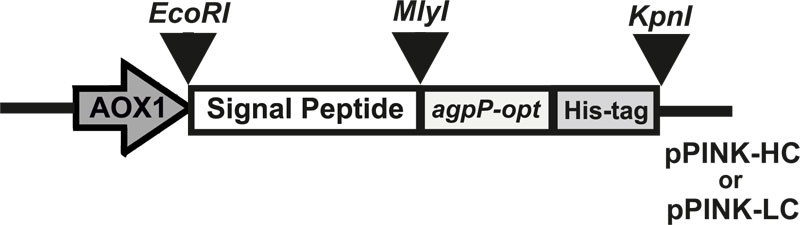
E. coli strain DH5α (Invitrogen, USA) was used for the propagation of recombinant constructions. E. coli transformants were selected on LB plates (100 μg/ml ampicillin). P. pastoris strain PichiaPink 4 (ade2, pep4, prb1) (Invitrogen, USA) was used as an expression strain and was grown on yeast extract peptone dextrose (YPD) medium (1% yeast extract, 1% glucose, 1% bactopeptone). Yeast transformants were selected on the Pichia adenine dropout (PAD) medium (1.34% Yeast Nitrogen Base (YNB), 0.125% Complete Supplement Mixture without adenine (CSM-ADE), 0.0005% biotin, 2% agarose, 100 ml 10Х dextrose). The buffered glycerol-complex (BMGY) medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer (рH 6.0), 1.34% YNB, 0.00004% biotin, 1% glycerol) and buffered methanol-complex (BMMY) medium (1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer (рH 6.0), 1.34% YNB, 0.00004% biotin, 0.5% methanol) were used for expression of recombinant proteins. Recombinant plasmid pUC57-agpP, containing codon-optimized gene sequence of histidine acid phytase from Pantoea sp. 3.5.1 (agpP) (GenBank accession no. KJ783401.1), was purchased from Genscript (USA). Plasmids pPINK-HC (high copy plasmid) and pPINK-LC (low copy plasmid) (Invitrogen, USA) were used for expression. Four different signal peptide sequences were examined: signal sequence of Aspergillus niger α-amylase gene, Aspergillus awamori glucoamylase gene, Gallus gallus lysozyme gene, and presequence of inulinase gene from Kluyveromyces maxianus.
2.2. Construction of P. Pastoris Strains Harboring agpP-opt gene
The gene sequence of histidine acid phytase from Pantoea sp. 3.5.1 – agpP (GenBank accession no. KJ783401.1) was used. For cloning and expression in P. pastoris, bacterial phytase gene sequence was changed: the native signal peptides sequences were excluded; C-terminal six-histidine tag sequence was added to the structural regions of the agpP and codon-optimization of nucleotide sequence was carried out. The Codon-optimized gene was synthesized and cloned into the pUC57 vector by the Genscript Company (USA). The optimized agpP gene was amplified from the pUC57-agpP plasmid by PCR using Phusion HF DNA polymerase (New England Biolabs, USA), an up-stream primer 5’-Ph-gccgctgatggcgatatgc-3’ (with blunt phosphorylated 5’-end) and down-stream primer 5’-agcggataacaatttcacacagga-3’ (with 3’ KpnI restriction site) using C1000 Touch thermal cycler (Bio-Rad, USA). The amplified agpP PCR product was digested with KpnI restriction enzyme (Thermo Scientific, USA). The pPINK-HC and pPINK-LC plasmids were digested with KpnI and EcoRI restriction enzymes (Thermo Scientific, USA). Signal peptide sequences of α-amylase, glucoamylase and lysozyme genes, and presequence of inulinase gene contained phosphorylated 5’-end with EcoRI restriction site for insertion into the pPINK-HC and pPINK-LC vectors and blunt phosphorylated 3’-end. Three-way ligation of agpP insert, plasmid (pPINK-HC / pPINK-LC) and signal peptide sequence (α-amylase / inulinase / glucoamylase / lysozyme) were carried out using Т4 DNA ligase (Thermo Fisher Scientific, USA).
E. coli DH5α cells were transformed with the resulting expression vectors [19], selected on LB plates (100 μg/ml ampicillin) and analyzed by colony-PCR using an upstream primer 5’-gactggttccaattgacaagc-3’ and a down-stream primer 5’-gcgtgaatgtaagcgtgac-3’(primers to AOX1 promoter and CYC1 terminator). The correct construction of expression vectors was verified by sequencing.
P. pastoris PichiaPink strain 4 was transformed with SpeI-digested expression vectors by electroporation according to the manual of the Pichia Expression kit (Invitrogen, USA). Electrocompetent yeast cells were prepared using a modified standard method [20]. Electroporated by Gene Pulser XcellTM Electroporator system (Bio-Rad, USA) yeast cells were then cultured on PAD medium plates and incubated at 30°C for 3-10 days. The integration of the expression construction into the yeast genome was analyzed by colony-PCR using primers to the AOX1 promoter and to the CYC1 terminator. Successful integration was checked by sequencing.
2.3. Expression of Recombinant AgpP-P Phytase
The P. pastoris selected clones were grown in 10 ml BMGY medium using 50 ml baffled flasks at 28°C for 24 h at 300 rpm shaking until the culture reached OD600 of 2-6. This 10 mL culture was then used to inoculate 200 ml of BMGY in 1 L baffled flask and was incubated at 28°C and 300 rpm shaking until the culture reached log-phase growth (OD595 = 2-6). The cells were harvested by centrifuging in sterile centrifuge bottles at 3000×g for 5 minutes at 28°C. To induce expression, the supernatant was decanted and the cell pellet was resuspended in 50 mL of BMMY medium using 250 ml baffled flask. The culture was incubated at 28°C and 300 rpm shaking for 72 h and induced by adding 1% (v/v) methanol at every 24 h intervals. Cells were harvested from the culture medium by centrifugation for 20 min at 3000×g at 4°C. The cell-free culture medium was analyzed for enzymatic activity and Lowry protein assay [21], gel electrophoresis (SDS-PAGE) and Western blotting.
2.4. Phytase Activity Assay
Phytase activity was quantified by the Greiner method measuring the amount of released inorganic phosphorous [22]. The reaction mixture contained 210μL 10мМ sodium phytate from rice (SigmaAldrich, Germany) in 100 mM sodium acetate buffer, pH 4.5 and 10-50 μL enzyme solution. The reaction was carried out at 37°C for 30 min and terminated by adding 900μL of freshly prepared 2:1:1 AAM solution (acetone–5N H2SO4–10 mM ammonium molybdate). After incubation for 2 min, 60μL of 1M citric acid was added to the reaction mixture. Blanks were prepared by adding AAM solution prior to the enzyme addition. Optical density was measured at 355 nm on a 2550 Microplate Reader (Bio-Rad, USA). A calibration curve was built using the concentrations of inorganic phosphate in the range of 5 to 600 nmol. One unit (U) of phytase activity was defined as the amount of enzyme required to liberate 1 μmol of inorganic phosphorous from sodium phytate per min at 37°C. Statistical significance was determined using a Student’s two-tailed t-test with significance set at a P-value of 0.05.
2.5. Protein Estimation, SDS-PAGE and Western Blotting Analyses
Protein concentration was determined by the Lowry assay using bovine serum albumin as a standard [21]. SDS-PAGE (12.5%) was performed, as described by Laemmli [23]. After electrophoresis, the gel was stained with Coomassie Brilliant Blue R-250 reagent and then destained with 10% acetic acid. Western blotting was performed by transferring the proteins from the non-stained SDS-PAGE to polyvinylidenedifluoride (PVDF) membrane (Sigma-Aldrich, Germany). After blocking, the membrane was incubated with the primary antibody – 6x-His Epitope Tag Monoclonal Antibody (HIS.H8) (Thermo Scientific), diluted 1:5000 prior to application. Afterwards, the membrane was washed with phosphate-buffered saline (PBS) and incubated with the secondary antibody – Pierce Goat Anti-Mouse IgG, (H+L), peroxidase-conjugated (Thermo Scientific), diluted 1:10000 prior to application. His-tagged proteins were detected with the SuperSignal West Pico Stable Peroxidase Solution and SuperSignal West Pico Luminol/ Enhancer Solution (Thermo Scientific).
2.6. Isolation and Purification of AgpP-P Phytase
Recombinant phytase AgpP-P was obtained from the cell-free culture medium of transformed P. pastoris culture induced with methanol for 36 h. Cells were removed from the culture medium by centrifugation for 20 min at 3000×g at 4°C. The resulting supernatant was passed through a 0.45 μm filter (Corning, Germany), pH of the supernatant was adjusted to 7.0. Purification of the recombinant phytase was carried out on Ni-NTA agarose (Qiagen, Germany). Before loading the supernatant, the Ni-NTA agarose-column was pre-equilibrated with buffer A (50mM Tris-HCl, 500 mM NaCl, 5% glycerol, 1mM PMSF, 2mM β-mercaptoethanol, pH 8.0). Serial washes were performed with buffer B (buffer A+ 0.1% triton X-100), C (buffer A+ 1M NaCl) and D (buffer A+ 10mM imidazole). Protein was finally eluted with buffer E (buffer A supplemented with 250mM imidazole). The eluted fractions were analyzed by phytase activity and Lowry protein assays, Coomassie blue-stained SDS-PAGE, and used for further biochemical assays.
Natural AgpP phytase of Pantoea sp. 3.5.1 was isolated and purified as described before [24] and used in comparative studies of biochemical properties of natural and recombinant enzymes.
2.7. Biochemical Characterization of Phytase
To determine the pH optimum of the enzyme, a standard phytase assay was performed in pH ranging from 1.0 to 9.0. Enzyme activity was measured in the following buffers at 100 mM final concentration: glycine-HCl (pH1.0 to 3.5), sodium acetate (pH 3.5 to 6.0), Tris-acetic acid (pH6.0 to 7.0), Tris-HCl (pH pH 7.0 to 9.0). To study pH stability, the enzyme was incubated in buffer solutions with different pH values at 4°C for 1 h, and the remaining enzyme activity was evaluated by a standard assay.
To study the temperature optimum of the enzyme, a standard phytase assay was performed at various temperatures ranging from 10°C to 70°C. To determine thermostability, the enzyme was first incubated at 10°C to 70°C for 1 h and cooled to 4°C, followed by measuring phytase activity under standard conditions.
The effect of divalent metal ions (Mn2+, Zn2+, Fe2+, Ca2+, Mg2+, Co2+) on enzyme activity was analyzed by preincubating the enzyme with each cation at 1mM final concentration.
The data are the means ± the standard deviations of at least three independent assays.
2.8. N-deglycosylation Analysis
N-deglycosylation of AgpP was carried out using Endo H deglycosylase (New England Biolabs). The reaction mix containing 1-20 µg of purified AgpP, 1 µl of 10X Glycoprotein Denaturing Buffer and H2O was incubated at 100°C for 10 minutes for denaturation of phytase. To the reaction mix, 2 µl of 10X G5 Reaction Buffer and 1-5 µl Endo H and H2O (to make a 20 µl total reaction volume) were added. The incubation temperature was 37°C for 1 hour. The N-deglycosylated proteins were analyzed by 12.5% SDS-PAGE.
2.9. Fermentation
P. pastoris pPINK-HC-inulinase-agpP strain was cultured in a Hinton flask containing BMGY at 30°C until OD595 value 10. 310 ml of the seed culture was added into a 5-l jar fermenter (Bioengineering RALF, China) containing 3.1 l of BMGY. The temperature and pH during fermentation were maintained constant at 30°C and 6.0, respectively. The stirring speed was set to 800 rpm. This batch culture was grown until the glycerol is completely consumed, which is indicated by a sharp increase in the level of dissolved oxygen (DO) to 100%. Once all the glycerol is consumed from the batch growth phase, a glycerol feed containing 50% w/v glycerol with 12 mL PTM1 trace salts per liter of glycerol was initiated to increase the cell biomass. The feed rate was set to 10 mL/hr /liter initial fermentation volume. After 6 h of glycerol feed, methanol induction was initiated by starting a 100% methanol feed containing 12 mL PTM1 trace salts per liter of methanol. The feed rate was set to 3.6 mL/hr per liter initial fermentation volume and was increased every 2 hours to 7.3 and 10.9 mL/hr/liter, respectively, and maintained at this level until the end of the fermentation process. Samples were taken periodically throughout the fermentation process for analyses of phytase activity and biomass.
3. RESULTS AND DISCUSSION
3.1. Construction of Recombinant P. Pastoris Strains
One of the advantages of extracellular protein expression in P. pastoris is the simple purification procedure of target protein because of the low level of endogenous proteins secreted by the yeast [25]. Target protein secretion efficiency depends on the secretion signal in the expression host as well as any native signal peptide that may be present in the recombinant protein [26]. In Pantoea sp. 3.5.1, phytase AgpP is localized in the periplasm of the bacteria and has 24 amino acid signal peptides [24]. For effective expression in P. pastoris, the sequence of phytase agpP (GenBank accession no. KJ783401.1) was optimized: the sequence of natural signal peptide was excluded, C-terminal His-tag was added in order to identify the enzyme by immune blotting and accelerate protein purification, and codon-optimization was performed.
Codon-optimization was carried out by GenScript (USA). OptimumGeneTM algorithm optimizes a lot of parameters that are important to the efficiency of gene expression, such as: codon usage bias, GC content, CpG dinucleotides content, mRNA secondary structure, cryptic splicing sites, premature PolyA sites, internal chi sites and ribosomal binding sites, negative CpG islands, RNA instability motif (ARE), repeat sequences, restriction sites that may interfere with cloning and others (http://www.genscript.com/codon-opt.html). Furthermore, it is known that codon optimization increases the expression of recombinant proteins up to 10-fold [27]. The synthesized 1765-bp agpP-opt gene showed 78% homology with the native agpP gene (GenBank accession no. KJ783401.1). All codons in agpP-opt were optimized for methylotrophic yeast P. pastoris.
Eight expression constructs were generated by inserting agpP-opt gene downstream of the secretion signal (α-amylase, inulinase, glucoamylase, or lysozyme secretion signal from Aspergillus niger, Kluyveromyces maxianus, Aspergillus awamori and Gallus gallus respectively) in pPINK-HC or pPINK-LC vectors (Fig. 1). These signal peptide sequences were shown to increase secretion of recombinant proteins in yeasts but have not been previously reported of so towards bacterial histidine acid phytase expression in P. pastoris.
The agpP-opt sequence was cloned into EcoRI and KpnI restriction sites of P. pastoris expression vectors pPINK-HC and pPINK-LC. The recombinant plasmids carried the agpP-expression cassette consisting of 1765 bp agpP-opt gene in frame with secretion signals, flanked by AOX1 promoter and terminator sequences. DNA sequencing of the final vectors confirmed the entirety of the agpP-opt gene sequence with added His-tag, in accordance with the agpP sequence from Pantoea sp. 3.5.1 [24]. Linearized plasmids were transformed into protease-deficient and ADE2 auxotroph P. pastoris PichiaPink strain 4 (ade2, pep4, prb1). PichiaPinkTM Strain 4 is a knock-out for proteinases A and B (i.e., pep4 and prb1), so, it has the lowest extracellular protease activity among all the PichiaPinkTM strains. The successful heterologous recombination and cloning of agpP-expression cassette into the host genome were confirmed by genotyping using 5’-AOX1 and 3’-CYC1 primers. PCR products of the expected size (about 1.7 kb) in all pPINK-agpP transformed P. pastoris cells denoted the integration of bacterial phytase gene into the yeast genome.
3.2. Expression of Recombinant AgpP-P in P. Pastoris
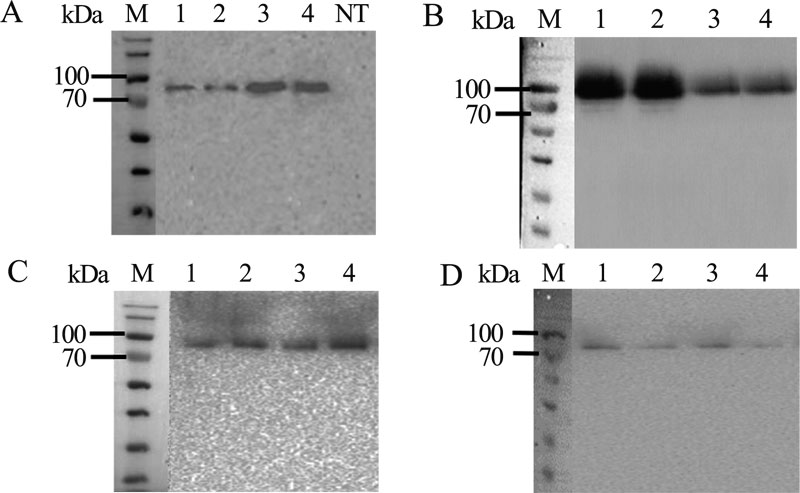
The pPINK-agpP transformed P. pastoris colonies were screened for complementation of adenine autotrophy, and for extracellular phytase activity. Two transformants with the highest extracellular phytase activity levels harboring each of the expression constructs (pPINK-HC-amyl-agpP, pPINK-LC-amyl-agpP, pPINK-HC-inul-agpP, pPINK-LC-inul-agpP pPINK-HC-glucoam-agpP, pPINK-LC-glucoam-agpP, pPINK-HC-lysoz-agpP, pPINK-LC-lysoz-agpP) were selected for shake flask expression and Western blot analysis (Figs. 2, 3).
After 36 h of methanol induction, extracellular and intracellular phytase activity was analyzed. All 16 clones exhibited extracellular phytase activity. Histidine acid phytase expression level in all transformants was low detectable in the intracellular lysates (less than 0.2 U/ml). Extracellular phytase activity in P. pastoris strain with an integrated construct based on pPINK-HC vector and Kluyveromyces maxianus inulinase gene signal sequence was almost twice higher than in other transformants – 3.2 U/ml. This activity of pPINK-HC-inul-agpP strain was 128-fold higher than the activity of the native Pantoea sp. strain 3.5.1 (0.025 U/ml) [24] and higher than in other expression systems: 17.7-fold higher than in E.coli pET28a-agpP (0.18 U/ml) [28] and 16-fold higher than in Yarrowia lipolytica pINA1296-agpP (0.2 U/ml) [29]. Taken together, all studied signal peptides were effective in the extracellular secretion of AgpP-P. Nevertheless, the combination of pPINK-HC vector and Kluyveromyces maxianus inulinase gene signal sequence showed the best result in phytase secretion by P.pastoris. This transformant was selected for further work.
Analysis of the expression level, due to determine the induction profile of recombinant AgpP-P phytase over time, revealed that the optimal time post-induction to harvest is 36 hours, after which phytase activity decreases, possibly due to protein degradation (Fig. 4A). Proteolytic degradation of secreted recombinant proteins may be caused by the destruction of the cell membrane when cells lyse during cultivation. Proteases, which are generally intracellular, are found in the fermentation broth of P. pastoris culture during high cell density fermentation. It is known that methanol is required to induce AOX1 promoter for recombinant protein production, although it itself makes up the conditions that launch increased production level of proteases and cell lysis, which leads to proteolysis of the secreted protein during high-density cultivation. Proteolytic activity rises during the induction process. Thus, it is necessary to evaluate the optimum induction time for recombinant protein secretion. The whole mechanism of the increase in protease activity in the culture broth is unknown, but methanol as carbon source leads to release or misdirection of proteases of vacuolar origin to the fermentation medium, inducing the degradation of secreted proteins [30].
In our study, we have made an attempt to produce AgpP-P phytase at a bioreactor scale (Fig. 4B). The fermentation process consisted of three phases. Phase I, initial cell growth and biomass accumulation on BMGY medium with glycerol, lasted 22 hours. The culture was grown until the glycerol is completely consumed, which is indicated by a sharp increase in the level of dissolved oxygen (DO). To ensure biomass growth, phase II was initiated by feeding additional glycerol. For such glycerol-fed batch culture, OD600 was around 40, and wet biomass was around 115 g/l. After 6 h of glycerol feed, phase III (induction) was initiated by feeding methanol. The maximum level of AgpP-P phytase secretion was detected at the 77th hour of the fermentation process. The yield of total extracellular phytase activity reached 4.38 U/ml. Currently, though our bioprocess has not yet been optimized, we produce recombinant thermostable phytase AgpP-P using a 5-liter bioreactor yielding up to 14000 U of phytase activity in the cell-free culture medium. Recombinant AgpP-P phytase is produced with a specific activity of 0.38 U/mg, product yield of 14000 U, with volumetric productivity of 56.8 U/ l /h. The bioreactor specific productivity of 1.6 U/ g/ h and biomass yield of 88 g/l were obtained.
3.3. Purification and Properties of Recombinant AgpP-P
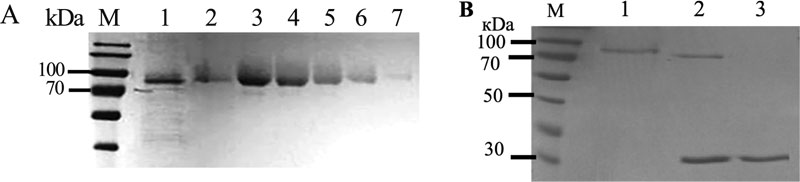
To fully characterize biochemical properties of the recombinant AgpP-P phytase, we isolated and purified the enzyme from P. pastoris culture liquid. We employed a one-step Fast Flow affinity chromatography protocol (Table 1). Cells were collected after 36 h of induction with methanol, and the culture liquid was filtered and passed over the Ni-Sepharose column. Protein was finally eluted with the buffer supplemented with 250mM imidazole. This resulted in a purification of 13.3-fold and an activity yield of 61.5%. Purified AgpP-P had a specific activity of 3.46 U/mg. This one-step protocol allowed us to purify the AgpP-P phytase to homogeneity (Fig. 5A).

| Purification step | Total protein (mg) | Total phytase activity (U) | Specific activity (U/mg) | Purification (fold) | Yield (%) |
|---|---|---|---|---|---|
| Culture liquid | 656 | 174 | 0.26 | 1 | 100 |
| Affinity chromatography | 31 | 107 | 3.45 | 13.3 | 61.5 |
The molecular weight of the native phytase was calculated to be 60800 Da and 61663 Da for His-tagged phytase according to the amino acid sequence. After SDS-PAGE separation, one single protein band with a molecular mass of nearly 80 kDa was observed (Fig. 5B, lane 1).
The NetNGlyC 1.0 Server (http://www.cbs.dtu.dk/servi ces/NetNGlyc/) predicted six potential N-glycosylation sites for AgpP-P (residues N55, N92, N106, N120, N234 and N570). However, it was not clear if the recombinant protein will be N-glycosylated in P. pastoris. SDS-PAGE analysis of AgpP-P after deglycosylation by Endo H showed a single band with a molecular size of 70 kDa (Fig. 5B, lane 2), verifying that AgpP-P is indeed N-glycosylated.
The pH optimum of AgpP-P phytase was studied in the range from 1.0 to 9.0. AgpP-P phytase exhibited activity within a pH range of 2.0 to 7.0 with its maximum at pH 4.0 (Fig. 6A), which corresponds to the pH-optimum of natural AgpP phytase of Pantoea sp. 3.5.1 (AgpP-N). More than 80% of activity was observed in a pH range of 2.0 to 4.0, AgpP-N showed only trace levels of activity below pH 3.0.

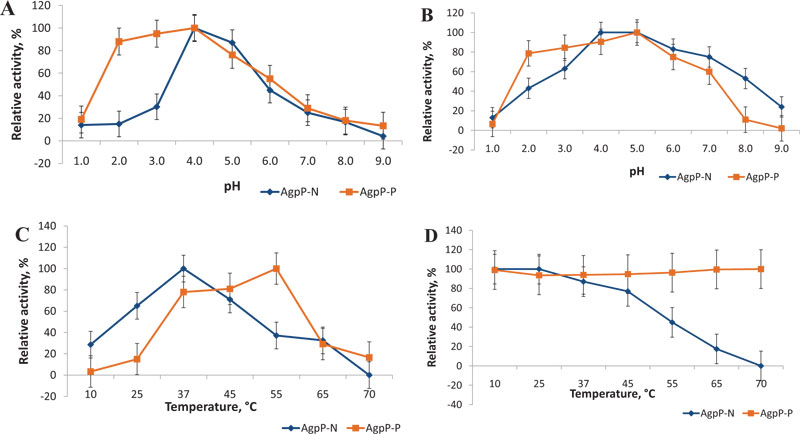
The effect of pH on enzyme stability was studied by incubating the enzyme for 1 h in a pH range of 1.0 to 9.0 at 4°C. AgpP-P remained very stable (more than 80% of activity) in the pH range of 2.0 to 4.0 in comparison with native phytase (AgpP-N), which displayed a sharper decrease in relative activity at pH below 4.0 (Fig. 6B). Enhanced levels of AgpP-P phytase activity and stability in acidic pH present in the forestomach (pH 4-5), gizzard and proventriculus (pH 2−5) of gastrointestinal tract of poultry are highly desirable properties for phytases used as feed additives [3]. This increase in pH stability of the recombinant phytase may be due to glycosylation, which is carried out by the host P. pastoris cells. It is known that glycosylation is a crucial posttranslational modification that can alter proper folding, functions and features of the protein [31, 32].
It was shown that histidine acid phosphatase (HAP) from Yersinia, expressed in Pichia pastoris and N-glycosylated, was more stable at the acidic pH of the poultry stomach than its unglycosylated form [33]. N-glycan at N124 played an important role in the adaptation to acid pH values and stability of Aspergillus fumigatus xylanase (Af-XYNA) heterologously produced in Pichia pastoris [34]. The pH stability was artificially engineered in penicillin G acylase, so the half-life of the glycosylated protein was 13-fold higher at pH 3 and 7-fold higher at pH 10 in comparison to its unglycosylated form [35].
The effect of temperature on the AgpP-P phytase activity was measured in the temperature range from 10°C to 70°C. The phytase showed a single temperature optimum at 50°C, which differs from that in native phytase (AgpP-N) (37°C) (Fig. 6C). Nevertheless, recombinant AgpP-P phytase showed high levels of phytate-degrading activity in the temperature range between 37-50°C, which overlaps with the internal body temperature of chicken (40-43°C).
As long as the feed pelleting process is usually carried out at high temperatures between 60 and 95°C, the thermal stability of the feed enzyme is a critical issue. Although phytase addition through the after-spray procedure helps to overcome potential heat destruction of the feed enzymes, thermostable phytases can often be a better candidate for feed supplements [3]. Natural AgpP phytase (AgpP-N) possessed stability for 1 h in the range of 10-45°C; higher temperatures caused enzyme inactivation [24]. Whereas, phytase activity of AgpP-P remained high even after 1 h of incubation at 70°C – more than 90% of the initial activity (Fig. 6D).
The thermal stability profiles of these two enzymes (wild-type and recombinant) were different, which is due to the fact that the recombinant phytase went through N-glycosylation by P. pastoris, while the native phytase from Pantoea sp. 3.5.1 was unglycosylated. N-glycosylation of recombinant proteins is considered to be a stabilizing factor and thus increases the thermostability of glycosylated proteins. Studies have shown the importance of attached carbohydrate chains and their role in the stability of glycoproteins [31]. The N-glycosylation enhances the structural rigidity of the proteins and causes a considerable decrease in dynamic fluctuations throughout the molecule, which leads to increased thermal stability [32]. There is a large number of studies reporting the increase in the thermal stability of proteins after glycosylation [36]. There are several bacterial phytases with improved biochemical properties after glycosylation as well [37]. For example, Bacillus phytase (PhyC) produced by P. pastoris was also glycosylated and showed higher residual activity, compared to non-glycosylated form, after 10 min at 70°C and 80°C [17]. Recombinant AppA phytase from Shigella sp. CD2 produced by P. pastoris showed enhanced thermostability of the protein after glycosilation – it retained around 70% of activity after 30 min at 70°C [18].
Glycosylation can lead to pH and temperature stabilization because of an increase in the internal electrostatic interactions of the protein. Glycans act as molecular spacers making the effective distance between the protein electrostatics and the solvent electrostatics bigger, which increases the strength of the internal electrostatic interactions for the protein, resulting overall in the enhanced conformational stability for glycosylated proteins [36].
Nevertheless, improved thermostability of recombinant AgpP-P phytase we observed is much higher than most known examples – phytase remains stable at 100% level (± 2%) after 1 hour of heat treatment at 70°C. Such high thermostability of the recombinant AgpP-P phytase has a clear industrial advantage.
To enrich poultry rations, many microelements are introduced as premixes, most often in inorganic form. It is known that metal ions are able to affect the activity of phytases and their activity may be inhibited. To study the influence of divalent metal ions on the recombinant phytase activity, we used Ca2+, Mg2+, Mn2+, Zn2+, Fe2+, Cu2+, and Co2+ ions at a 1 mM concentration (Table 2). Fe2+, Cu2+, and Zn2+ ions inhibit AgpP-P activity by 40%, 45%, and 40%, respectively. Inhibition of phytase activity can possibly be connected not only with the action of ions in the active center of the enzyme but also with the formation of insoluble chelates with the substrate and making it out of reach for hydrolysis by phytase [38]. On the other hand, Ca2+, Mg2+, and Mn2+ ions increase enzymatic activity over 2-fold. The activity and stability of beta-propeller phytases from the Bacillus genus are increased in the presence of Ca2+ [39]. Similarly, the activity of AppA phytase from Shigella sp. CD2 was enhanced in the presence of Ca2+, Mg2+ and Mn2+ [18]. Thus, metal ions can have the same profile of action on both recombinant and native phytases. Positive regulation of the AgpP phytase activity by some of the metal ions can be beneficial in comparison with the traditional industrial phytases.
| Metal ions | Relative activity (%) | |
|---|---|---|
| AgpP-N | AgpP-P | |
| Control | 100 | 100 |
| Ca2+ | 230±16 | 220±15 |
| Mg2+ | 218±14 | 200±14 |
| Mn2+ | 215±14 | 200±14 |
| Co2+ | 100±7 | 98±7 |
| Fe2+ | 90±6 | 40±3 |
| Cu2+ | 84±5 | 45±3 |
| Zn2+ | 58±4 | 40±3 |
CONCLUSION
The recombinant form of the histidine acid phytase from Pantoea sp. 3.5.1 produced in P. pastoris had similar biochemical properties to those of the native form, except for increased thermal stability. The pH-optimum in the acidic pH and the temperature-optimum at internal body temperature of chicken as well as resistant to thermal denaturation give advantages to AgpP-P for the diversification of phytases implementation as a feed additive. Overall, the enhanced thermostability of the glycosylated AgpP-P phytase, its high-yield expression provided by strong inducible promotor and effectively designed expression cassette, the simple purification procedure of the secreted enzyme, and the possibility of large-scale expression make the foundation for further production of this bacterial phytase in P. pastoris at an industrial scale.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of this research are available in the manuscript.
FUNDING
This work was supported by the Russian Science Foundation (Project No. 16-16-04062) and was performed in accordance with the Russian Government Program of Competitive Growth of Kazan Federal University Kazan, Russia.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Authors would like to specially thank Ph.D Rafil Khairullin for help in protein fermentation and purification.

