All published articles of this journal are available on ScienceDirect.

Integrating Microbiome Network: Establishing Linkages Between Plants, Microbes and Human Health
Authors Info & Affiliations
Abstract
The trillions of microbes that colonize and live around us govern the health of both plants and animals through a cascade of direct and indirect mechanisms. Understanding of this enormous and largely untapped microbial diversity has been the focus of microbial research from the past few decades or so. Amidst the advancements in sequencing technologies, significant progress has been made to taxonomically and functionally catalogue these microbes and also to establish their exact role in the health and disease state. In comparison to the human microbiome, plants are also surrounded by a vast diversity of microbes that form complex ecological communities that affect plant growth and health through collective metabolic activities and interactions. This plant microbiome has a substantial influence on human health and environment via its passage through the nasal route and digestive tract and is responsible for changing our gut microbiome. This review primarily focused on the advances and challenges in microbiome research at the interface of plant and human, and role of microbiome at different compartments of the body’s ecosystems along with their correlation to health and diseases. This review also highlighted the potential therapies in modulating the gut microbiota and technologies for studying the microbiome.
1. INTRODUCTION
Although, evolved independently, the plant roots and animal guts both serve similar functions of nutrient uptake, provide metabolic capabilities to host organisms, regulate gene expression and offer protection against pathogens [1-4]. Animal guts and plant roots both converge in harboring diverse and complex microbial classes of bacteria [5, 6], archaea [7, 8], fungi and oomycetes [3], viruses [3, 9] as well as eukaryotes [10, 11]. Unraveling the diversity along with molecular functions of different microbiomes associated with plants, animals and the human health and how these microbiomes work across the cross-kingdom, has become a major challenge for scientists. However, the availability of high throughput sequencing technologies has enabled the scientific community to taxonomically and functionally characterize this unseen majority of microbes up to a much greater extent [12]. Analyzing these microbiomes by employing the Next-Generation Sequencing (NGS) capabilities has helped researchers to reveal the vast majority of microbial diversity associated with plants, animals and humans [4, 13]. NGS, along with other molecular techniques has greatly expanded and deepened the extent of microbiome research by uncovering novel ways to understand the microbial world, disease etiology and a number of other scientific studies [1, 14], A rapid increase in microbiome rese- arch entails analyzing an adequate number of samples directly from the environmental sites using appropriate sequencing technology that helps in resolving the rare species or to perceive subtle variations within the collected microbial samples. Recently, a number of review papers have been published, where the authors have stressed either on plant or human microbiome [1, 4], however, in the present review the authors have discussed the microbiome studies at the interface of plants and humans by establishing a linkage between the two. This review primarily focused on the advances and challenges in microbiome research at the interface of plants and humans, the role of microbiome at different compartments of the body’s ecosystems along with their correlation to health and diseases. This review also highlighted the potential therapies in modulating the gut microbiota and technologies for studying microbiome. Moreover, the present review discussed the linkages between the microbes, plants and humans by emphasizing on the concept of herbivore.
2. MICROBIOME AT THE INTERFACE OF PLANT AND HUMAN HEALTH
Plants and animals, including humans, are universally and persistently colonized by microbes and these microbes are intimately co-evolved with their host-organisms, influencing their functioning and evolution [15]. Plant and animal hosts provide a multitude of ecological niches for microbes, allowing these diverse microbial classes to coexist and to form microbial communities. Globally, plant and animal scientists are increasingly recognizing the impact of these microbial communities on their host organisms [16]. As microbiome encompasses hundred-times more genes than their host genomes, hologenome (host genome and its microbiome) can fluctuate with respect to time and space [17]. Microbiotas can act as a phenotypic plastic buffer between the two determining factors (host-genotype and environmental effects) responsible for shaping the host phenotype. Thus, the microbiome interactions with the host and surrounding environment are the key determinants of the host phenotype and this phenotypic expression of the host organism largely depends on the presence and composition of the associated microbes [18]. Similar to the human microbiome, plant microbiome is also one of the essential determinants of plant health and productivity [13]. Additionally, the studies carried out previously [19], suggested the essential roles of plant microbiome in shaping phenotypic, epigenetic plasticity and evolutionary conservation of plants.
Microbes have shaped and continue to shape the environment from billions of years [20]. Microbial communities offer a multitude of functions for the interacting organisms, thereby altering host development, physiology, fitness and wellbeing. Elucidating how the microbiome affects the fitness and well being of the host organisms has turned out to be a major challenge for the researchers. On the one hand, phyto-hormones regulate the physiology of plants and also architect the plant microbiome [2], however, on the other hand, the pathogenic, symbiotic and commensal microbes produce and imitate plant hormones to change their plant hosts along with the microbial communities. Although less appreciated, animals, including the humans and microbes are believed to produce and recognize phytohormone and more remarkably, these hormones are recognized to affect cellular processes, inflammatory responses and glucose homeostasis [21]. This, in turn, has significant implications for human health. Studies carried out by Chanclud and Lacombe [22] suggested that the plant hormones, produced by gut microbiota or obtained from the diet, impact the human health (Fig. 1). These suggestions were supported by studies [23], where their results showed the impact of plant hormones on human diseases such as Inflammatory Bowel Disease (IBD), diabetes and cancer, which are also modulated by the gut microbiome. The plant hormones are generally ingested in the form of the diet, with consequences for human health; however, the mode of action of these hormones is unclear. In the near future, the outcomes of omics research related to the human gut microbiome need to be exploited to answer these questions. Studies conducted by different researchers [15, 24, 25] suggested that, experience gained from the plant microbiome may prompt the animal microbiome studies and vice versa. Some studies suggested that humans are gaining health benefits from various plant colonizing microbes either directly by food or indirectly by breathing [26-28]; hence, this area is recently under severe investigation. One of the important reasons for how this microbiome inside the gut changes, is the food that we take. The fermented food is highly rich in useful microbial content, which is an attractive “functional food” [29]. The link between the diversity of microbes and food has also implemented to preserve food [30, 31]. There are some microbes that naturally protect different foods such as they preserve vegetables from rotting. However, during the storage of food, the microbial profile often gets changed [32, 33]. Wall et al. [34], in their study, mentioned that the microbial diversity inhabiting different soils plays an immense and imperative role in maintaining crop growth and finally, human health. Studies carried out by Kish et al. [35], revealed that the Bacillus Calmette Guerin (BCG) or a mycobacterium vaccination offers many health benefits other than providing some protection against tuberculosis. Interestingly, many Gram-negative microbes are unable to produce spores, therefore, most of them share similar strategies for survival (dormancy, by producing potent osmolytes, phase variation, etc.), and these strategies facilitate their perseverance in the environment [36]. The domestication of humans, plants and animals in the course of time has resulted in the domestication of their allied microbiomes [37, 38]. One of the best examples of artificial (man-made) co-evolution is the reduction of glucosinolates in Brassicaceae, which is bitter in taste via breeding. Glucosinolates provide protection to plants against different pathogens (e.g., Verticillium in oilseed rape); moreover, their metabolites contain anti-tumor activities in humans [39].
3. HUMAN MICROBIOME: OPENING THE BLACK BOX
When a baby is born, a new island appears in the microbial space. Humans provide the microbes a wonderfully rich and diverse habitat ranging from UV-exposed skin to dark, anoxic energy-rich gut. There is a need to understand and answer challenging questions for the future, such as how our association with microbes has evolved, forces that shape it, how this co-evolution impacts our health and how the changes in the biosphere may affect it. Humans bear an unusual impact on the environment, and the environment also influences our exceptionally diverse gut microbiota across the globe. Until now, these properties of human microbiota were mainly seen as a “black box” [13]. The in vitro, cultivation, which served as the cornerstone of the traditional microbiology till the 1970s, cannot be used for most of the densely colonized microbial communities [14, 23]. However, the development of the science of metagenomics and high throughput sequencing technologies has improved our horizons by generating a huge amount of the data sets that can be excavated for gathering the information regarding the type composition and functional properties of enormous microbial communities. Studies regarding the human microbiome have now reached a critical inflection point, where, we are shifting from the description and exploration for understanding the mechanism of action and to develop novel clinical interventions on the basis of these understandings [40]. These advances have created a major shift in the translational research resulting in significant private investment, in terms of the so-called big-pharma. This move towards the clinical microbiome studies is supported by advancements in the personalized medicine, e.g., the rapid reduction in the cost of DNA sequencing is allowing accurate and rapid identification of the precise treatment procedures, thus allowing a positive outcome of the diseases such as type 2 diabetes, cancer and neurological disorders [41]. There are several key challenges in microbiome analyses, particularly in defining the differences between two samples from unrelated or related individuals or from the same individual, between different time scales, or in terms of the evolutionary biology of different species. Once these differences are defined, statistical methods can be developed to analyze the data and establish correlation and phylogenetic distance metrics to compare microbial communities using 16S rRNA or 18S rRNA genes to allow phylogenetic classification of metagenomic samples. Here in Table 1, we compiled the research conducted on the microbiome in different organs and species using sequencing technology.
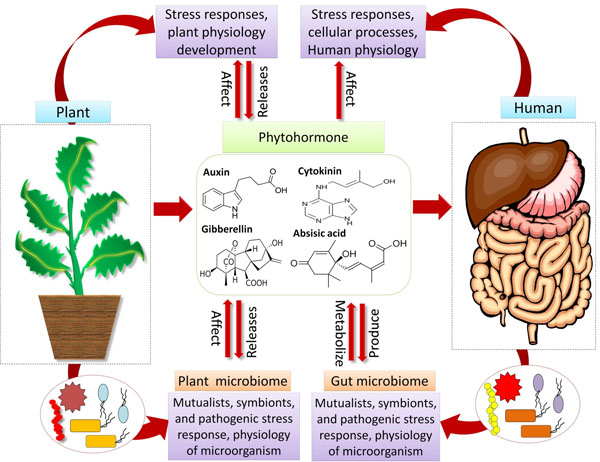
Table 1.
| S. No. | Host | Sample Source | Sequencing Method | Amount of Data Retrieved | References |
|---|---|---|---|---|---|
| 1. | Rhinopithecus bieti | Feces | 454 pyrosequencing | 97,942 pyrosequencing reads | [77] |
| 2. | Elephant | Feces | 16S rRNA-based sequencing | 454 reads | [78] |
| 3. | Phascolarctos cinereus | Hindgut | 16S rRNApyrosequencing | 81,608 | [79] |
| 4. | Human | Vagina | 16S rRNA-based sequencing | 2.5 million reads | [80] |
| 5. | Human | Gut | Illumina-based metagenomic sequencing | 576.7 gigabases of sequence | [81] |
| 6. | Dog | Feces | 454 pyrosequencing | 201,642 reads | [82] |
| 7. | Cow | Rumen | Whole genome sequencing | 268 G of metagenomic DNA | [83] |
| 8. | Cat | Feces | 454 pyrosequencing | 187,396 reads | [82] |
| 9. | Human | Feces | 16S rRNA-based sequencing | 9773 16S rRNA sequences | [84] |
| 10. | Mouse | Feces | 16S rRNA-based sequencing | 4172 16S rRNA sequences | [85] |
| 11. | Mouse | Cecum and feces | 16S rRNA-based sequencing | 2878 16S rRNA sequences | [86] |
| 12. | Human | Feces | 16S rRNA-based | 2064 16S rRNA sequences | [87] |
| 13. | Mouse | Cecum | 16S rRNA-based sequencing | 5545 16S rRNA sequences | [88] |
| 14. | Zebrafish | Intestine | 16S rRNA-based sequencing | 5545 16S rRNA sequences | [88] |
| 15. | Human | Colonic mucosa and Faeces |
16S rRNA-based sequencing | 11,831 16S rRNA sequences | [89] |
| 16. | Mouse | Cecum | 16S rRNA-based sequencing | 5088 16S rRNA sequences | [90] |
4. THE BODY'S ECOSYSTEM AND MICROBIOME FUNCTIONS
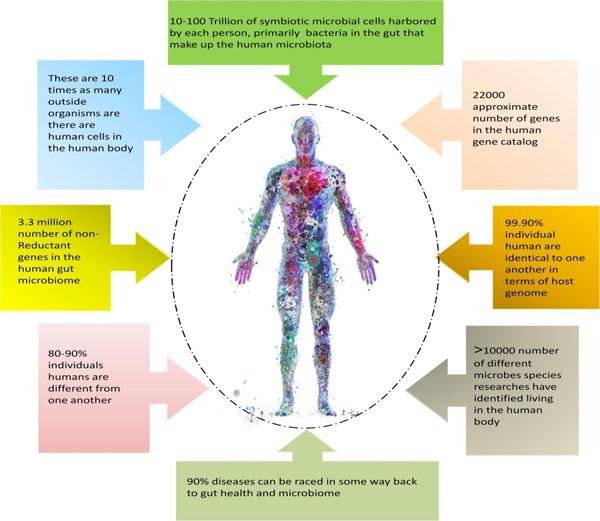
The microbiome composition and function differ according to age, sex, location, race and diet of the individual [42]. The microbiome composition also shows individual specificity, with the differences being much larger than the typical biochemical differences that occur within an individual over a given time period [43]. The relative contribution of the lifestyle, genetics, diet and environmental factors to the composition of the human microbiome is still unclear [13, 44]. As already mentioned, the human body is teemed with trillions of microbes, the commensal bacteria that reside on and inside the human body outnumber the body's own cells. Similar to other living organisms, humans too, are the result of a continuous association between the cells of diverse origins and genetic pedigree, integrating the members of all the three domains (Eukarya, Bacteria and Archaea) of life [45-47]. The microbes colonizing different organs of the human body lead to the development of human-associated microbiomes (Fig. 2). Significant biotic and abiotic gradients do exist in the body that led to microbial compartmentalization. Diverse microbial communities subside the human body, and have a fundamental role in human health. Studies carried out by Rutayisire et al. [48] and Franasiak and Scott [49] revealed that the microbiome of every healthy individual exhibits remarkable differences in species composition. Within the body, the microbe colonizes different parts such as skin, oral and nasal cavity, gut, intestines, reproductive tract and perform diverse functions, forming an appropriate ecosystem within the body. Ensuring the long- term and enduring contribution of these microbiomes to good health, and physiology may necessitate more than transient enrichments with specific microbial communities and metabolites. Good health requires an incessant cross-talk between the microbes and their host in a symbiotic relationship [50]. In humans, the maximum diversity of microbes is found in the gut, where they synthesize the essential nutrients such as amino acids and vitamins, which are required for performing various body functions. Like, humans, the plants are also colonized by diverse microbial communities, herbivory provides a cross-kingdom (animal and plant) microbiome link, where the microbes are transferred from the plant and are established in the gut. Individual studies carried out by different workers [51, 52], suggested that microbes being the part of our diet can either improve human health or can cause infectious outbreaks by transferring the pathogens.
Microbes associated with the human body function in a synergistic way with body cells and influence the outcomes of health both across the lifespan and generations. This microbiome performs its actions by influencing different pathways such as neural, endocrine and immunologic. While the bulk of microbes flourish in the oral cavity, urogenital tract and on the skin, however, those colonizing the gut are the most diverse and abundant and their functions are believed to be best understood [1].
Right from birth, the gut microbiome is of profound importance for an organism's health, as it helps in maintaining the homeostasis of the GI tract. The gut microbiome serves as a barrier against the pathogenic microbes and plays a vital role in the digestion and metabolism of a variety of food materials [53]. The microbiome also plays an active role in breaking different toxins and drugs, besides the synthesis of vitamins and ion-absorption and also produces immunomodulatory metabolites and maintains the immune system. The microbiome also bears a profound effect on the development of the host immune system and physiology. A number of diseases (such as IBD, MALT lymphoma, stomach cancer, necrotizing enter colitis and obesity) both in infants and adults are found to have links to the microbiome [23, 54]. These analyses show prospects for future extraordinary findings and the correlation of microbiomes with their influences on different aspects of human biology. Therefore, there are comparatively few examples that describe the mechanism regarding how these microbes interact and lead to the exploitation of this knowledge. We are still not aware of what defines a healthy microbiome. Most microbiota and their related diseases depend on microbial profiling or metagenomics, but these studies are not applicable to describe the mechanisms involved [55]. However, in the future, two related areas of research will directly mark these limitations. Interactions between microbial populations and their hosts, particularly humans, are necessary to understand the development and functionality of the host. In this regard, metagenomic studies are gaining increasing attention. The National Institutes of Health started as a far-reaching Human Microbiome Project to identify microbial species that live at different sites of the human body and to observe their function in regulating digestion and metabolism and evaluated their impact on immunity. The outcomes of these studies may play a significant role in improving our understanding of the influence of the microbiome on an individual’s health and disease and therefore, aid in developing personalized medicine approaches for the future. Metabolomics in microbiome studies was considered the first area that was already well researched [56]. A magnificent example of this particular validation is that of butyrate, which is a short-chain fatty acid often secreted by particular Clostridial species, which interact with G-protein-coupled receptors (e.g., GPR41 and GPR43) that modulate the regulatory T cells. Therefore, it is clearly evident how these approaches can be exploited to alter disease and improve health. However, we are not very aware of the metabolites that are generally secreted by these microbes. Hence, these are considered vital molecules through which microbes communicate with their host or with each other. The potential to grow these specific microbes and the communities of microbes in the culture will permit the identification of these molecules. This approach was integrated with biological experiments e.g. the human microbiome transplanted into mice by the expression of the immune system of humans. This will make new tools available to study the interference of communication between the microorganism and host [53].
5. POTENTIAL THERAPIES AIMED AT MODULATING THE GUT MICROBIOTA
Different types of interaction between the host and microbes may cause many diseases and often lead to the disruption of homeostatic cooperation. Various drug discoverers and therapists now largely depend on therapies that are target-specific as well as on the ecology of these microbes to recognize, understand and estimate the after-effects of these treatments [57, 58]. The ramifications of these microbially targeted therapies often alter the composition of communities by wiping out peculiar strains of specific species. The secondary infections that are related to the use of antibiotics provide an advisory report of the accidental ramifications of disconcerting a network of microbial species. Apprehension regarding microbial ecology will assist the advancement of probiotics; therefore, their beneficial anticipations will rely on meticulous impersonal trials. Probiotic is a term mainly used to name bacteria that are associated with beneficial effects for both humans and animals. These probiotics will likely be taken in the form of the consortia of distant future residents of the community: a fecal transplant in a capsule [59]. The members of the genera Lactobacillus and Bifidobacterium are mainly but not exclusively used as probiotic microorganisms. Fecal microbiota transplantation, one of the well-known therapies, is a process in which stool from a healthy donor is placed into another patient’s intestine [60]. Several studies pertaining to the role of gut microbiota in protecting human health have been conducted and have justified their positive role [61, 62]. The study conducted by Buffie and Pamer [63] has validated the therapeutic potential of gut microbiota when manipulated as probiotics and genetically engineered commensals. There are many success stories related to fecal transplantation conducted by different scientists for the treatment of different diseases such as recurrent Clostridium difficile-associated diarrhea [64], Parkinson’s disease [65], inflammatory bowel diseases [66], obesity [67], recurrent Clostridium difficile symptomatic infection [68], alcoholic hepatitis [69], etc. The study conduc- ted by Mayor [70] depicted that the Fecal transplantation is used for recurrent or difficult-to-treat Clostridium difficile symptomatic infection. As the intestines contain hundreds of different types of bacteria, however, when the proportion of the healthy bacteria decreases, more often because of the antibiotic use, the harmful C. difficile bacteria proliferate and cause diarrhea. Though some of the antibiotics treat C. difficile, however, some individuals do not respond to these antibiotics; in that case, the patients can be treated through stool transplant. Transplanting the stool from healthy donors restores the balance of healthy bacteria and help in clearing the infection. The potency of microbiome-targeted therapies will need to be appraised by employing novel diagnostic devices and tools that amplify the functions of communities instead of measuring their composition, in addition to the materialistic response to the communities of microbes for perturbation such as with a probiotic or an antibiotic.
6. MICROBIOME SIGNATURE AS BIOMARKERS: ROLE IN HUMAN HEALTH AND DISEASES
The concept of biomarkers is essential to expand the knowledge regarding environmental studies, which may help in understanding the link between environmental exposure and the symptoms of the disease in exposed populations. The microbes that are site-specific, need to encounter the xenobiotics before their absorption across the skin, gut the respiratory system. Published literature has revealed that these microbiomes participate in biotransformation and interact with the “niche”, and therefore, transduce the responses both to and from the host organism. The integration of the microbiome into the environmental health paradigms will broaden and amplify our concepts of susceptibility, along with the xenobiotic interactions and other factors that affect the status and function of these barrier systems [71]. The microbiome associated biomarkers are known to play an important role in the studies of metabolism and its outcomes such as autoimmune disease, asthma and metabolic syndrome [72].
7. HOST EFFECT ON THE PLANT MICROBIOME
The host-microbe interactions are extremely dynamic and complex, and are largely controlled by the status of the host and its surrounding environment. In one study, the mutants of Arabidopsis thaliana deficient in SAR (Systemic Acquired Resistance) showed variations in the rhizosphere microbial community composition as compared to that of the wild type, however, even the activation of SAR upon chemical treatment did not result in any significant change in rhizosphere microbial community. In A. thaliana, the induction of salicylic acid-mediated defense abridged the diversity of endophytic bacteria in the phyllosphere. However, the plants that are deficient in jasmonate-mediated defense exhibited elevated levels of epiphytic diversity. These studies propose that the effects of plant defense processes on microbial diversity are variable and that systemic acquired resistances are responsible for regulating populations of some bacteria. In many cases, plants release some chemical signals (flavonoids) that facilitate specific interactions and are perceived by other organisms [73]. In the case of mycorrhiza, strigolactones elevate hyphal branching and also stimulate seed germination of the parasitic plants. In many cases, the plant genes and pathways play an important role in developmental pathways, shared between rhizobial and mycorrhizal symbioses [74]. A number of antimicrobial compounds (alkaloids, phenolics and terpenoids) are produced by the plants and these compounds are widely distributed throughout the plant kingdom both in terms of its constitution and response to pathogens. Some chemical comp- ounds are confined to particular groups, e.g., glucosinolates, a group of secondary metabolites that are only produced by the members of order Brassicales [75]. Similarly, Arabidopsis produces glucosinolates and Avenastrigosa (oat) produces triterpenoid saponins. These studies highlight that even a minor change in plant genotype can induce an intricate and unanticipated effect on plant microbiome and hence, on disease resistance [76]. The role of plant microbiome and its relationships to productivity, health and biogeochemical cycles need to be considered as much as the plant itself.
8. TECHNOLOGIES FOR STUDYING MICROBIOME
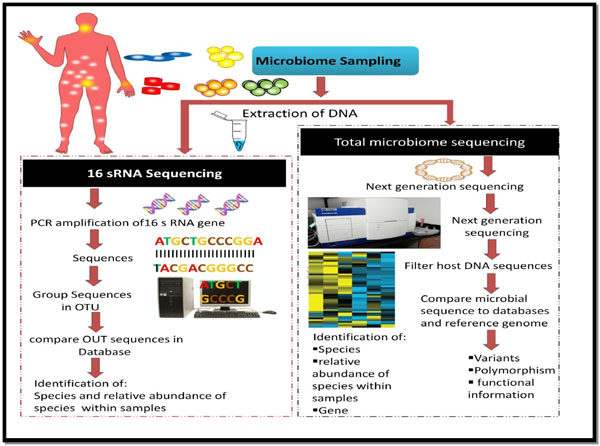
After successful DNA sequencing, the obtained sequences need to be analyzed and interpreted, so as to obtain meaningful information. The enormous amount of the data generated as a result of the next generation sequencing necessitates sophisticated analytical tools. A number of approaches do persist for analyzing the data. Some of the tools mentioned in this article are used for microbiome research. Owing to its novelty and advancement, high-throughput sequencing technology is rapidly advancing in its quality, speed and cost. At present, abundant data have been generated by next-generation sequencing globally; thus, there is an urgent need to store this large amount of data along with the analysis with great precision. Fig. (3) shows the schematic representation of procedures used in microbiome analysis. A number of tools available to analyze the metagenomic data are presented in Table 2, and discussed in detail in a review published by the authors [4].
| S. No | Software | Platform | Description | Website | References |
|---|---|---|---|---|---|
| 1. | CowPI | Stand alone | A functional inference software used for rumen microbiome analysis. | http://www.cowpi.org | [91] |
| 2. | Microbiome Analyst | Web server | A complete statistical, visual and meta-analysis software for microbiome data analysis. | http://www.microbiomeanalyst.ca/ | [92] |
| 3. | EBI | Web server | To compare functional analyses of sequence. | https://www.ebi.ac.uk/metagenomics/ | [93] |
| 4. | MetaBAT | Web server | Binning millions of contigs from thousands of samples. |
https://bitbucket.org/ berkeleylab/metabat |
[94] |
| 5. | GraPhlAn | Web server | Produces high-quality, compact visualizations of microbial genomes and metagenomes. | http://segatalab.cibio.unitn.it/tools/graphlan | [95] |
| 6. | deFUME | Web server | It is an east to use web server for trimming, assembly and functional annotation of sanger sequencing data derived from functional selection experiments. | http://www.cbs.dtu.dk/services/deFUME/ | [96] |
| 7. | METAVIR | Web server | Annotate viral metagenomics sequences (raw reads assembled contigs). | http://metavir-meb.univbpclermont.fr/ | [97] |
| 8. | VAMPS | Web server | An integrated tool used for data visualization and analysis obtained for microbial population. | https://vamps2.mbl.edu | [98] |
| 9. | MyTaxa | Web server | to identify the taxonomic affiliation of a query genome sequence or a sequence of a contig assembled from a metagenome, including short sequences (e.g., 100-1,000nt long), and to classify sequences representing novel taxa at three levels (whenever possible), i.e., species, genus and phylum. | http://enve-omics.ce.gatech.edu/mytaxa | [99] |
| 10. | Phinch | Web server | It provides an interactive visualization tool that allows users to explore and manipulate large biological datasets. | http://phinch.org/ | [100] |
| 11. | Orione, | Web server | Orione, a Galaxy-based framework consisting of publicly available research software and specifically designed pipelines to build complex, reproducible workflows for next-generation sequencing microbiology data analysis. | http://orione.crs4.it. | [101] |
| 12. | PICRUSt | Stand alone | A computational approach used for functional profiling of a metagenome using 16S amplicon sequencing. | http://picrust.github.com/ | [102] |
| 13. | FANTOM | Stand alone | Comparative analysis of metagenomics abundance data. | http://www.sysbio.se/Fantom/ | [103] |
| 14. | Meta Microbes Online | Web server | Offers phylogenetic analysis of genes from microbial genomes and metagenomes. | http://meta.MicrobesOnline.org | [104] |
| 15. | METAGENassist | Web server | METAGENassist is a freely available web server for comparative metagenomic analysis. Comparative metagenomic studies involve the large-scale comparison of genomic or taxonomic census data from bacterial samples across different environments. | http://www.metagenassist.ca/METAGENassist/ | [105] |
| 16. | HUMAnN | Stand alone | Analysis of metagenomic shotgun data from the Human Microbiome Project. |
http://huttenhower.sph.harvard.edu/ humann | [106] |
| 17. | MetaPhlAn | Stand alone | Faster profiling of the composition of microbial communities usingunique clade-specific marker genes. | http://huttenhower.sph.harvard.edu/metaphlan | [107] |
| 18. | MetaVelvet | Stand alone | High quality metagenomic assembler | http://metavelvet.dna.bio.keio.ac.jp/ | [108] |
| 19. | SOAPdenovo2 | Stand alone | Metagenomic assembler, specifically for Illumina GA short reads. | http://soap.genomics.org.cn/soapdenovo.html | [109] |
| 20. | WebMGA | Web server | A customizable web server for metagenomic analysis. | http://weizhongli-lab.org/metagenomic-analysis/ | [110] |
| 21. | CoMet | Web server | The CoMet-Universe server allows you to analyze the taxonomic and functional composition of your metagenomic sample and to compare it with a large collection of publicly available data from previous metagenome studies. | http://comet.gobics.de | [111] |
| 22. | MEGAN | Stand alone | Diversity analysis and visualization (needs similarity alignments as input). | http://ab.inf.uni-tuebingen.de/software/megan | [112] |
| 23. | QIIME | Stand alone | Data trimming and filtering, diversity analysis, and visualization. | http://qiime.org/ | [113] |
| 24. | Galaxy portal | Web based | Web repository of computational tools that can be run without informatic expertise. | https://usegalaxy.org/ | [1] |
| 25. | METAREP | Web server | Flexible comparative metagenomics framework. | http://www.jcvi.org/metarep/ | [114] |
| 26. | SmashCommunity | Stand alone | Performs assembly and gene prediction mainly for data from Sanger and 454 sequencing technologies. | http://www.bork.embl.de/software/smash/ | [115] |
| 27. | mothur | Stand alone | Fast processing of large sequence data. | http://www.mothur.org/ | [116] |
| 28. | RAMMCAP | Stand alone | Ultra fast sequence clustering and protein family annotation. | http://weizhonglab.ucsd.edu/rammcap/cgibin/rammcap.cgi | [117] |
| 29. | Phymm | Stand alone | Phylogenetic classification of metagenomic short reads usinginterpolated Markov models. | http://www.cbcb.umd.edu/software/phymm | [118] |
| 30. | MG-RAST | Web server | High-throughput pipeline for functional metagenomic analysis. | http://metagenomics.anl.gov/ | [119] |
| 31. | WebCARMA | Web server | Unassembled reads as short as 35 bp can be used for the taxonomic classification with less false positive prediction. | http://webcarma.cebitec.unibielefeld.de/ | [120] |
| 32. | CAMERA | Web server | Provides list of workflows for WGS data analysis. | https://portal.camera.calit2.net/gridsphere/gridsphere | [121] |
| 33. | CD-HIT | Web server | Identity-based clustering of sequences. | http://weizhonglab.ucsd.edu/cd-hit/ | [122] |
| 34. | TETRA | Web server | Correlation of tetra nucleotide usage patterns in DNA. | http://www.megx.net/tetra | [123] |
9. FUTURE PROSPECTS
A wealth of intact information occurs within the gut microbiota, providing key insights into microbial effects on host organisms, most of which are entirely unknown. In spite of the huge progress in the exploration of microbes at the interface of plant and human health, some more practical and fundamental studies are required to unravel the microbiome network. Future research on the microbiome may focus on the following:
- Microbiome editing for the diagnosis, prevention and treatment of diseases.
- Use of microbiome as a biomarker for disease diagnosis and treatment.
- To assess, whether the change in the microbiota, associated with health conditions is structure driven or function driven.
- Development of approaches and analysis pipelines for the elucidation of altered community function.
- Up till now, the microbiome studies have mainly focused on bacterial inhabitants, however, future research should focus on ascertaining the role of fungi, viruses, and other eukaryotes in the health and disease state of their hosts (plants and animals).
CONCLUSION
The study of microbes in relation to the plant and human health through the application of novel and advanced techniques has significantly enhanced our perceptive regarding the structure, specificity and function of below-ground (plant) and gut (animal) microbiome. Metagenomics, an applied field of research has broadened the prospects in the field of cultureless future of microbiology, environmental biotech- nology with potential uses in the on-site restoration of ecosystems and development of strategies for bioremediation, public health and food safety. Metagenomic tools, analysis software and databases are rapidly increasing in number; however, gaps persist in the analysis techniques and algorithms that sometimes limit, data mining. However, with the increased technological advancements, it is much easier to delineate the data, and hence the mechanisms by which microbes affect their hosts. Moreover, recognizing and understanding linkage among the microbes, plants and human health, will, in turn, help in understanding the disease etiology and ecosystem functioning. From here, we support that there exists a prodigious potential for the microbiome research to constantly design, execute and share novel studies, results and to realize, understand and comprehend the full potential of the enhanced outcomes over a wide range of human diseases and global change ecology.
AUTHOR'S CONTRIBUTIONS
Suresh Babu Naidu Krishna and Ashwani Kumar conceived the idea of this review and prepared the manuscript. Anamika Dubey and Muneer Ahmad Malla prepared the illustrations and tables. Richa Kothari, Chandrama Prakash Upadhyay and Jamila K Adam edited the manuscript.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Suresh Babu Naidu Krishna would like to thank DUT, Durban South Africa. Anamika Dubey would like to thank DST for financial supports in the form of DST inspire research Fellowship.

