All published articles of this journal are available on ScienceDirect.

Taxonomic and Functional Annotation of Termite Degraded Butea monosperma (Lam.) Kuntze (Flame of the Forest)
Abstract
Background:
Butea monosperma is an economically and medicinally important plant that grows all over India, however, the plant is highly susceptible to termite attack. The present study unravelled the bacterial community composition and their functional attributions from the termite degraded Butea.
Methods:
Total genomic DNA from termite degraded Butea monosperma samples was extracted and subjected to sequencing on Illumina's Miseq. The raw and unassembled reads obtained from high-throughput sequencing were used for taxonomic and functional profiling using different online and stand-alone softwares. Moreover, to ascertain the effect of different geographical locations and environmental factors, comparative analysis was performed using four other publically available metagenomes.
Results:
The higher abundance of Actinobacteria (21.27%), Proteobacteria (14.18%), Firmicutes (10.46%), and Bacteroidetes (4.11%) was found at the phylum level. The genus level was dominated by Bacillus (4.33%), Gemmatimonas (3.13%), Mycobacterium (1.82%), Acidimicrobium (1.69%), Thermoleophilum (1.23%), Nocardioides (1.44%), Terrimonas and Acidithermus (1.09%) and Clostridium (1.05%). Functional annotation of the termite degraded B. monosperma metagenome revealed a high abundance of ammonia oxidizers, sulfate reducers, dehalogenators, nitrate reducers, sulfide oxidizers, xylan degraders, nitrogen fixers and chitin degraders.
Conclusion:
The present study highlights the significance of the inherent microbiome of the degraded Butea shaping the microbial communities for effective degradation of biomass and different environmental toxicants. The unknown bacterial communities present in the sample can serve as enzyme sources for lignocelluloses degradation for biofuel production.
1. INTRODUCTION
Butea monosperma (Lam.) Kuntze is native to the tropical and sub-tropical parts of the Indian Subcontinent and Southeast Asia, and is distributed across India, Bangladesh, Nepal, Sri Lanka, Myanmar, Thailand, Laos, Cambodia, Vietnam, Malaysia, and western Indonesia. Butea is commonly known as ‘Flame of the forest’ and belongs to the family Fabaceae [1]. Locally, this plant is known as Palash, Palas, Mutthuga, Bijasneha, Khakara, Dhak Chichra, Bastard Teak, Bengal Kino, and Nourouc [2]. The crude extracts of various parts of this plant has been reported to have antifungal, anti-inflammatory, antibacterial and hypoglycemic activities. The plant has also been found to possess astringent, tonic, diuretics, and aphrodisiac properties. The widespread uses of Butea in traditional systems of medicine have resulted in their extensive chemical analysis for their bio-active components [3]. The plant holds a significant place because of its medicinal and other miscellaneous uses of economic importance [4]. Morphologically, the plant exhibits distinct features with large sized flowers that are arranged in a rigid racemes of 15 cm length. 3 flowers together form the tumid nodes of the dark olive-green velvety rachis. Flowers are used as vegetables by the tribal's and are also used for the preparation of dye for colouring garments and for making skin antiseptic ointments [4]. The above mentioned reasons make Butea an important and much sacred plant species.
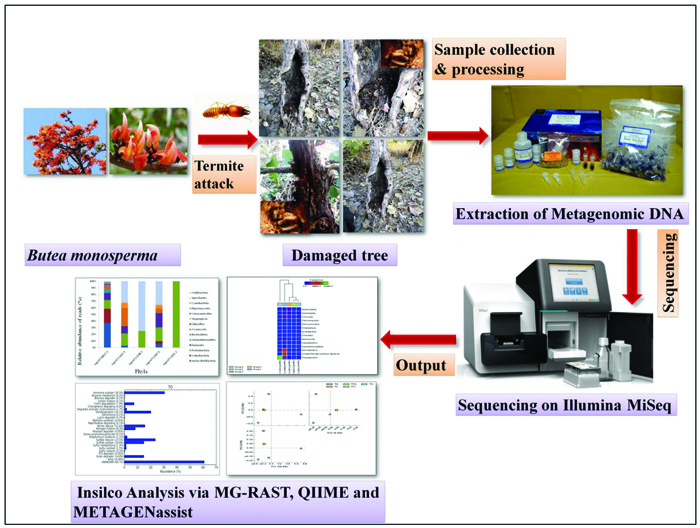
Termites are ubiquitous and major constituents of the forest ecosystem in the tropical and subtropical areas [5]. Apart from Antarctica, termites are found in almost all the continents [6] and are well known for their capacity to damage, and destroy wood and wooden products of human homes, building materials, forests, agriculture crops and other commercial products [7]. Termites, besides being small insects, have a successful evolutionary history mainly because of their sociality and capability to efficiently digest lignocellulose [8]. Presently, these tiny creatures contribute more than 95% of the insect biomass in the tropical soils [9]. About 3,000 species of termites have been described so far, however, comparatively only few species pose threat to wooden structures. The principal component of their diet is wood, which is also the dominant structural element of the building construction industry. Termites have the ability to degrade 65-99% and 5-83% of the wood-cellulose and hemicelluloses and lignin within 24 hours under natural conditions [10, 11]. In fact, termites possess the inimitable characteristics in order to handle lignin-carbohydrate complexes efficiently. A variety of lignocellulolytic enzymes do occur in the termite gut [12, 13]. Termites, in spite of their small body size, harbor an abundant and astonishingly diverse gut microbiome, which represents one of the most fascinating examples of symbiosis both within the microbes, as well as between animal and microbes [14]. It is widely accepted that lignocellulose digestion in termites is closely related to the host as well as the specific flora of symbiotic microbes [15, 16]. Various evidences do suggest that the lignin structure was modified in termites and that the modification process begins in the foregut and continues in the mid gut [17, 18]. From the perspectives of both basic research as well as biotechnology, there arises a substantial interest in accomplishing a clear perceptive of the diversity of biological mechanisms that are employed during lignocellulose degradation [19, 20]. Around the globe, termites comprise one of the extremely successful groups of organisms that are capable of degrading wood and are therefore vital for expanding their roles both in carbon turnover and as possible source of biocatalysts for the efforts that are aimed at converting the wood biomass into biofuels. The main aim of the present study is to unravel the structural composition and functional attributions of different microbial communities from the termite degraded B. monosperma and to study the variation in microbial community structure with respect to changing geographical locations and environmental factors. For this, we used the Next Generation Sequencing technology targeting the hypervariable region of the microbial 16S rRNA genes to unravel the bacterial community residing in termite degraded Butea sample. The complete work flow of this work is presented in Fig. (1).
2. MATERIALS AND METHODS
2.1. Sample Collection and Preparation
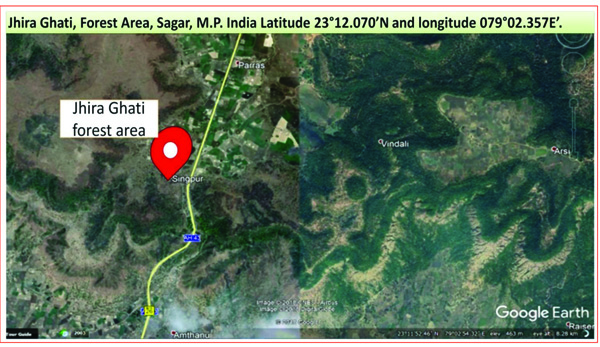
A total of nine samples from termites degraded Butea monosperma tree were randomly sampled from Jhira Ghati, Forest Area, Sagar, M.P. India (Latitude 23°12.070’N and longitude 079°02.357E’) as shown in Fig. (2). All the collected samples were properly labelled and packed in plastic bags, and were immediately transported to the laboratory and stored at 4°C till further use.
2.2. DNA Extraction and 16S rRNA Amplicon Sequencing
In order to profile the full-scale taxonomic diversity, the total bacterial genomic DNA was extracted from 250 mg/sample using the Mo Bio Power soil DNA isolation kit (MoBio Laboratories, Inc. CA, USA) as per the manufacturer’s instructions. The purity and integrity of the extracted genomic DNA were assessed through agarose gel electrophoresis (1%) and the concentration was quantified via NanoDrop 2000 spectrophotometer (Thermo Scientific) by measuring the absorbance at 260/280 nm. Total genomic DNA extracted from three replicates was pooled to one sample in order to cover the maximum diversity. The DNA samples were stored at -20°C till sequencing. Illumina’s MiSeq system was used for the 16S rRNA amplicon sequencing of extracted total genomic DNA as this instrument is a dominant platform for amplicon sequencing in microbial ecology studies due to its great flexibility, high-throughput, fast-turnaround time, longer sequence reads and higher accuracy.
2.3. Taxonomic Profiling of Termite 16S rRNA Gene Using MG-RAST
In this study, we have analysed the 16S rRNA gene sequences using Metagenomic Rapid Annotation Using Subsystem Technology (MG-RAST) server which is a comprehensive metagenome analysis server and offers a number of advantages like open access, publicly available metagenomes as well as comparative analysis of different metagenomes. The raw and unassembled sequences generated from high-throughput sequencing via Illumina were uploaded to MG-RAST server v 4.0.3 (http://metagenomics.anl.gov/) for taxonomic annotation and profiling under MG-RAST ID number mgm4738027.3 [21]. The low quality regions were trimmed using SolexaQA [22, 23] following downstream analysis by using RDP as the reference database with the parameters set as default.
2.4. Comparative Taxonomic and Functional Diversity for Selected samples by QIIME
QIIME is an open-source software pipeline built by making use of PyCogent toolkit [24] and is accessible at http://www.qiime.org/ or http://github.com/qiime/. For comparative taxonomic profiling and annotation of the termite metagenome with other termite metagenomes, the OTU table of the four publically available 16S rRNA amplicon metagenomes was downloaded from MG-RAST server v 4.0.3 (http://metagenomics.anl.gov/) [21] and analysis was carried out by QIIME release 1.9.1 using default parameters by Caporaso et al. [25]. The details of the four different geographically located termite metagenomes used for the comparison purposes are listed in Table 1. To ascertain functional profile of the termite degraded Butea monosperma, the taxonomic abundance dataset was uploaded on the METAGEN assist server [26] (http://www. metagen assist. ca/METAGENassist/) which performs taxonomy to phenotypic mapping.
3. RESULTS
3.1. Illumina Sequencing
There are various approaches to study the microbiota of the termite degraded Butea such as culture-independent and culture-depended. The culture dependent molecular approaches can trace very less amount of bacterial diversity [27, 28] whilst the culture independent methods still have some limitations such as they are time consuming, costly, low coverage and insufficient to reflect the true bacterial diversity, however recent advances in molecular approach of high throughput next-generation sequencing technology have provided large scale analysis with unprecedented depths and coverage [29, 30]. Nowadays, the use of NGS technology has made it possible to study the descriptive metagenomics. Metagenomics study can reveal the community structure and variation of the microbiome and the relative microbial abundance is estimated based on different physiological and environmental conditions [31, 32]. Ultimately, these studies can help to reflect connections between the identity of a microbe or a community and their respective functions in the environment [33].
The total genomic DNA extracted from the microbial communities of the termite degraded Butea was subjected to Illumina sequencing for the bacterial 16S rRNA gene region (v3 and v4). Tagmentation (fragmentation and adapter tagging), PCR amplification and clean-up of the genomic DNA were performed as per Illumina Nextera library preparation guidelines (Illumina, USA). 4nM libraries were prepared, pooled and sequenced on the Illumina Miseq desktop sequencer (2×300 run). The reads generated were then subjected to quality check and were subsequently analyzed using the RDP database classifier with parameters set at default values [34].
3.2. Taxonomic Composition of the Bacterial Metagenomes from Termite Sample
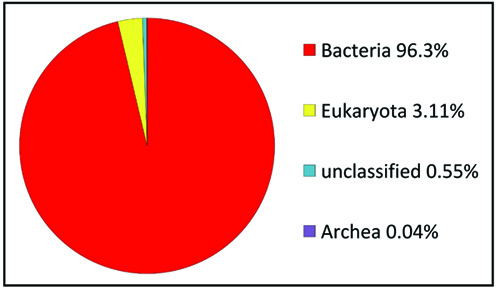
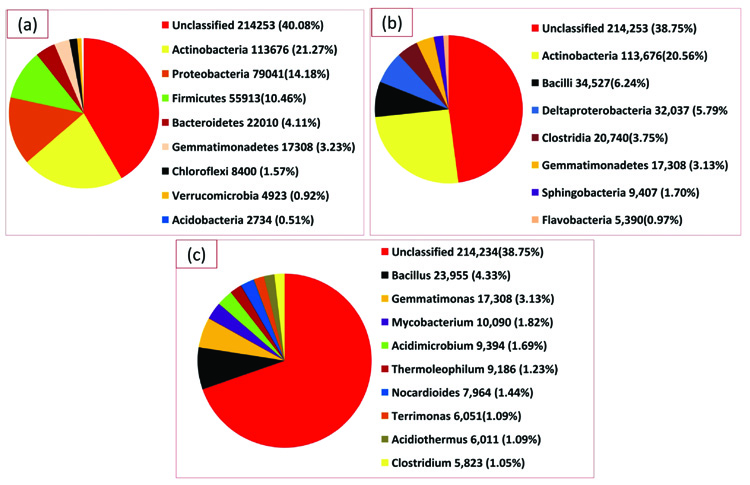
A total of 168,310 sequences, with 86,275,309 base pairs and an average length of 513 bps were obtained following the sequencing through Illumina sequencer of the termite degraded Butea sample (TO) (Table 2). The taxonomic profiling of the DNA samples extracted from the termite degraded Butea was performed against RDP database with the default parameters and the phylotypes were analyzed upto the genus level. The MG-RAST server used to assess the quality of this termite metagenome confirmed that 96.35% of the sequences belong to the bacteria, with 3.11% being Eukaroyata, followed by Archaea 0.04% and the unclassified being 0.55% (Fig. 3). The phylum level composition of the bacterial communities of the termite degraded Butea sample was composed of 9 phyla with one being unidentified. At the phylum level, Actinobacteria was predominant with a relative abundance of (21.27%) followed by Proteobacteria (14.18%), Firmicutes (10.46%), Bacteroidetes (4.11%) and Gemmatimonadetes (3.23%). The rest of the phyla showed a relatively minimum abundance with Chloroflexi showing a percentage abundance of (1.57%) followed by Verrucomicrobia (0.92%), and Acidobacteria (0.51%) (Fig. 4a). Level 3rd (class) distribution within these phyla can be observed in Fig. 4b. At the genus level, Bacillus were predominant, with the relative abundance of 4.33% followed by Gemmatimonas (3.13%), Mycobacterium (1.82%), Verticillium (1.70%), Acidimicrobium (1.69%), Thermoleophilum (1.23%), Nocardioides (1.44%), Terrimonas and Acidithermus (1.09) and Clostridium (1.05%) (Fig. 4c).
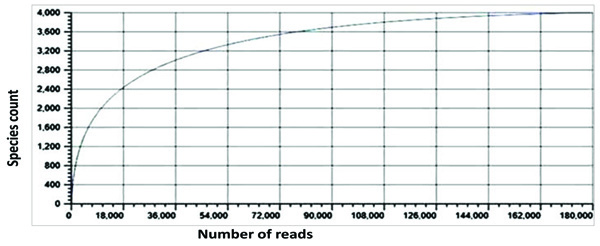
3.3. Rarefaction Analysis
Rarefaction curve is an index used to estimate the sampling depth. The adequacy of the sampling was estimated by means of rarefaction curve. The sampling efforts and the species richness of the termite degraded Butea sample were estimated by means of the rarefaction analysis. The current results depict that the rarefaction curves for this sample reached saturation, which corresponded to the true bacterial diversity within the sample (Fig. 5).
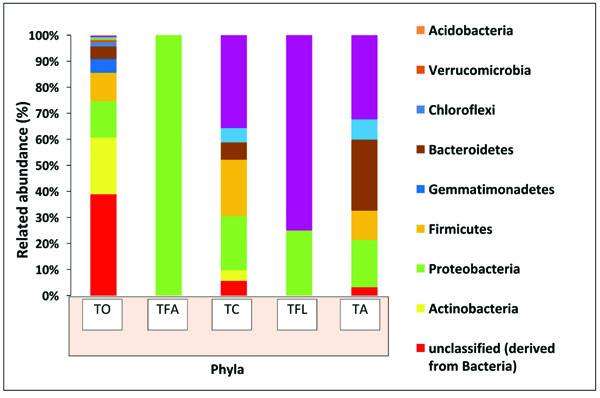
3.4. Comparative Taxonomic Profiling of Termite Degraded Butea with Other Publically Available Termite Metagenome Samples
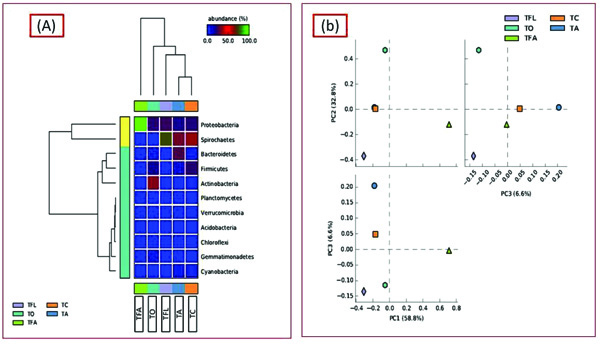
In order to ascertain the variation in microbial diversity with respect to different geographical location and environmental conditions, the termite degraded Butea sample (TO) was then compared with four other publically available termite degraded plants metagenomes. The four different termite degraded plant metagenomes used for comparison purposes were (TA), (TFL), (TC) and (TFA) from Australia, la Coubre France, Soroa Cuba and Aledo lA France respectively, with each belonging to different geographical location and having different weather conditions. To our surprise, a varying trend was observed at the phylum level with Actinobacteria dominating in the present study (TO) while Bacteroidetes dominating in TA and Firmicutes in case of TC. Notably, Proteobacteria is the second most abundant taxa in three samples (TO, TA, TC) whereas the same (Proteobacteria) is the most dominant in case of TFL and TFA (Fig. 6). Comparative taxonomic profiles of the bacterial metagenomes from termite degraded Butea (TO) and other termite samples (TA, TC, TFL and TFA) at the phylum level using MG-RAST based-RDP database indicate a significant variation in the relative abundance for bacterial composition between different termite samples (Fig. 7a). The PCA plot generated illustrated the clustering of the metagenomes as individual samples, thereby indicting a significant variation in microbial composition within these termite metagenomes. The PCA plot of the 5 termite metagenomes using the taxonomic classification at phylum level showed that the termite metagenome (TO) did not cluster with the termite samples (TA, TC, TFL and TFA), which more or less cluster with each other. The present metagenomics study of the termite had significantly higher Actinobacterial but a much lesser Bacteroidetal reads compared to TA (Fig. 7b). Nonetheless, the termite metagenome from
| S.NO | MG-RAST ID | Symbol/Code | Microbiome | Location | References |
|---|---|---|---|---|---|
| 1. | mgm4738027.3 | TO | Termite metagenome | Sagar, (M.P) India | Present Study |
| 2. | mgm4711607.3 | TA | Termite gut microbiota | Darwin, NT, Australia | [47] |
| 3. | mgm4711598.3 | TFL | Termite gut microbiota | la Coubre, France | [47] |
| 4. | mgm4711587.3 | TC | Termite gut microbiota | Soroa, Cuba | [47] |
| 5. | mgm4711601.3 | TFA | Termite gut microbiota | Aledo l A, France | [47] |
| Sequencing Method | Library Used | Pre QC | Post QC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| bp Count | Sequences Count | Mean Sequence Length | Mean GC Percent | bp Count | Sequences Count | Mean Sequence Length | Mean GC Percent | ||
| Sequencing by synthesis | Paired-end | 86,275,309 | 168,310 | 513 ± 26 bp | 60 ± 4% | 24,073,023 | 46,757 | 515 ± 20 bp | 60 ± 3% |
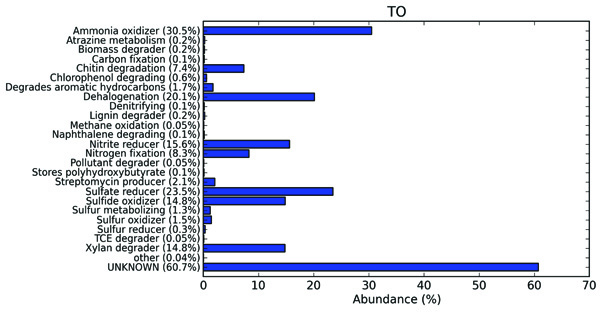
3.5. Taxonomic to Phenotypic Mapping of the Termite Degrade B. Monosperma Metagenome
The extensive in silico analysis of the termite degraded B. monosperma metagenomic data by METAGENassist, a web based server, provides taxonomic phenotypic mappings of the datasets. The functional annotation was carried out by taking into consideration the metabolic compositions (Fig. 8). The functional mapping of the sample illustrated that termite gut can be viewed as a micro-ecological habitat, thereby serving as an environment for a number of bio-geochemical cycles. The representation of the functional abundance in the termite degraded B. monosperma showed a significance of the processes such as ammonia oxidation, chitin degradation, xylan degradation, nitrate reduction, nitrogen fixation, dehalogenation and sulfate reduction. The occurrence of these processes gives the indications of the operation of the sulphur and nitrogen cycles with in this micro-environment. In the present sample, the relative abundance of the different groups is as ammonia oxidizers (30.5%), sulfate reducers (23.5%), dehalogenators (20.1%), Nitrate reducers (15.6%), sulfide oxidizers (14.8%), xylan degraders (14.8%), nitrogen fixers (8.3%), and chitin degraders (7.4%), with a lower percentage abundance of sulphur metabolizing, lignin degraders and pollutant degraders.
4. DISCUSSION
Metagenomic studies provide a deeper knowledge of the complex microbial consortia inhabiting any environment [35]. Assessment of the bacterial diversity using amplicon sequencing relies on the hyper-variable regions of 16S rRNA. The aim of the present study was to ascertain taxonomic composition of the termite degraded Butea using 16S rRNA amplicon sequencing. In order to obtain a meaningful data from termite sequencing, the data obtained from the present study was compared with the termite sequences that were available publically, so as to get an insight of the termite microbiome.
The study of normal gut microbiota is essential as species composition and proportion changes with respect to varying environmental conditions. The extensive use of high throughput next generation sequencing helps to unravel the impact of external factors. Till now, there is very less knowledge regarding the biodiversity analysis of termite degraded Butea using HT-NGS. In our study, we used the 16S rDNA amplicon study of the termite degraded Butea microbiota and compared our results with the publically available termite degraded plants to characterize the bacterial diversity in termite gut and to investigate the diversity and species abundance variation with changing environmental conditions and geographical locations. As the gut microbiota can be influenced by a number of factors including diet, environment and locations, our data presented in this study should be considered as location-specific.
The most of the predominant genera found in our study are common residents of termite gut [36]. Taxonomic profiling and categorization of the bacterial communities predicted that termite gut environment harbors a diverse array of bacterial communities that are unique to those of other environments [37]. In the present study, Actinobacteria dominated (21.27) out of the classified sequences (≥62%) followed by Protobacteria, Firmicutes, Gemmatimonadetes, Bacteroidetes, Chloroflexi, Verrucomicrobia, Planctomycetes and Cyanobacteria. Similarly, a different trend was observed in the bacterial diversity of other samples (Fig. 6). The variation in bacterial diversity may be associated with the change in environmental conditions (like pH, temperature, oxygen content, altitude and feeding habit) because the colonization of the microbes relies on the environmental factors [38, 39]. Protobacteria are usually characterized by their dominance in early stages of composting of plant materials and hence are responsible for cellulose degradation [40, 41]. Thus, the colonization of Protobacteria can be endorsed with their ability to hydrolyze the hemi-cellulose. The abundance of bacterial phyla Actinobacteria (TO), Bacteroidetes (TA), Spirochaetes (TFL), Proteobacteria (TFA) and Firmicutes (TC) has shown their contribution to symbiotic relation of the termite species, acetogenesis (acetate production) and nitrogen acquisition, and cellulose degradation [38, 42]. Termite microbiome incorporates abundant and morphologically distinct communities of Spirochetes, which own metabolic properties such as N2 fixation and H2/CO2-acetogenesis, thereby contributing to the nitrogen, carbon as well as energy requirements for the termite hosts. The presence of Spirochaetes, although relative lesser in abundance in the termite degraded Butea metagenome signifies that the plant is a good source of carbon and nitrogen and also reflects diverse degradation and bioconversion mechanisms and pathways. The presence of Acidobacteria (although relatively much lesser in abundance) in our sample (TO) and its absence in other samples hints towards the acidic nature of the environment. In the same way, their PCA is also differentiated by similar spread pattern. The STAMP generated PCA plot illustrated the clustering of the termite degraded Butea and other termite metagenomes as unique samples. Analysis revealed that the samples entirely had different bacterial compositions which may be due to varied dietary habitats as well as different geographical locations and environmental conditions (pH, carbon, nitrogen, oxygen, and temperature) [43]. This seems to be a foreseeable result as other studies hold the same view [44]. As environmental conditions can directly alter the physiological status of the endemic microbial communities [45, 46]. Here, we conclude that the shift within the microbial diversity to a large extent depends on environmental conditions.
CONCLUSION
After sequencing on Illumina platform and comparative analysis of termite degraded Butea collected from forest area of Sagar, M.P. India (TO), the results obtained indicate that this study is ideal for the taxonomic profiling of different bacterial communities responsible for the degradation of Butea. Taxonomic profiling of the termite metagenome showed that Actinobacteria was dominant followed by Proteobacteria, Firmicutes, Gemmatimonadetes, Bacteroidetes. Preponderance of the Protobacteria suggests that termite is the chief degrader of lignocellulose on the Butea. This study is the first of its kind to explore the taxonomic diversity of the termite responsible for the degradation of the Butea. The microbial community is influenced by various environmental factors (moisture and increase in temperature) within the degraded pile of Butea. As the location and environmental factors change, so does the bacterial diversity. A number of the sequences from the sample are only classified upto the domain level or either the phylum level, signifying the presence of these uncultivable or novel bacterial species. The isolation of the bacteria from the termite degraded Butea monosperma with diverse functions such as nitrogen cycling and dinitrogen fixation, production of hydrolytic enzymes, and pollutant degradation may prove to be of great importance to industries and scientific communities that are focused towards the degradation of ligno-cellulosic biomass for biofuel production. To develop approaches in order to cultivate these unknown and novel bacteria sets a big stage for utilizing these novel organisms for various biotechnological applications.
AUTHOR'S CONTRIBUTION STATEMENT
AK executed the experimental work, designed and wrote the manuscript. MM and AD analysed and arranged the data, PV gave some suggestions.
ETHICAL APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
UGC-BSR Start-up grant by University Grant Commission, New Delhi, India.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
AK would like to acknowledge UGC-BSR Start-up grant by University Grant Commission, New Delhi, India for providing financial support for this study. AD would like to acknowledge the Department of Science and Technology, New Delhi, India for providing financial support through the DST Inspire Ph.D. Fellowship (IF160797).

