All published articles of this journal are available on ScienceDirect.

Microbiota of a Full-scale UASB Reactor Treating Brewery Wastewater Using Illumina MiSeq Sequencing
Authors Info & Affiliations
Abstract
Background:
The efficiency of biological wastewater treatment plant is determined by bacterial metabolism. There are data on the effect of operational parameters on microbial consortia present in laboratory scale reactor. However, knowledge on the full-scale reactor is still limited at present, hence the need to define the relations between the microbial structure and the performance of full-scale reactor.
Objective:
In this study, the microbial community structure in a full-scale UASB reactor treating brewery wastewater was assessed using metagenomics Next-Generation Sequencing technique.
Method:
Granular sludge samples were collected from the UASB reactor treating brewery wastewater and extracted genomic DNA was amplified using barcoded bacterial primer sets targeting V3-V4 region of the 16S rRNA genes on sequencing Illumina MiSeq platform.
Results:
The taxonomic analysis revealed the abundance of bacteria (~95%) with considerable Archaea community (~2%) in the granular sludge. After trimming, 18 bacterial phyla, 29 orders, 36 families and 44 genera were recovered from the 48,488 sequences reads of the 16S rRNA genes analysed, where the most abundant community belongs to Firmicutes, Bacteroidetes, Synergistetes and Proteobacteria phyla.
Conclusion:
For a sustainable bioenergy generation, understanding the mechanisms of anaerobic system in relation to microbial community is an important factor to increase the production of biogas production during wastewater treatment. To the best of our knowledge, this report is one of the studies that explored and described bacterial diversity and community structure of a full-scale UASB reactor treating brewery wastewater using high-throughput sequencing. This study provides insight into the dominant microbial community and their phylogenetic diversity in biogas producing reactor.
1. INTRODUCTION
Brewery wastewater is high in organic content with high biodegradable values, but if it is not properly treated before being discharged into the environment, could lead to serious environmental problems. Due to this fact, biological wastewater treatment through the use of anaerobic digestion technology has become a very attractive method for the treatment of brewery effluent [1-5]. In recent times, symbiotic relationship between the microorganisms that are involved in the conversion of organic matter in the industrial wastewater and their activities to convert the complex organic matter to simple molecules and biogas generation has attracted much attention due to their low-tech nature and ability to operate onsite and reclaim water for reuse [6-8].
Technologies such as high rate Upflow Anaerobic Sludge Blanket (UASB), Expanded Granular Sludge Bed (EGSB) and Anaerobic Moving Bed Biofilm Reactors (AMBBR) offer an alternative to conventional treatment methods due to their ability to treat industrial wastewaters of high Organic Loading Rates (OLR) at a low Hydraulic Retention Time (HRT) with the potential of generating bioenergy [9-11]. A UASB reactor is among the most widely accepted high-rate anaerobic reactor that uses dense aggregates of granules as the core component during the treatment of different types of wastewater [12]. Anaerobic reactors are often considered as a black box due to an underestimation of microbial community present in them due to limited techniques that are available for the microbial identification. However, its performance depends on the function and structure of microorganisms present in the reactor [13]. Therefore, a deeper understanding of the microbial community in anaerobic digester treating different substrate is vital to develop new strategies that could be used in maintaining and improving the efficiency and stability of the treatment plant.
For a better understanding of microbial consortia in anaerobic digestion systems, several researchers have employed the use of culture-independent methods like clone library analyses, molecular fingerprinting or real-time PCR methods for identification and quantification purposes [14-18]. A good number of such studies focused on the composition and structure of microbial communities [6, 19], especially the archaeal and bacterial communities. However, it is also observed that the majority of the sequences analyzed were not assigned to an established genus in either communities [20, 21]. This indicates that few researches have been conducted on the identification of important key microbial communities in an anaerobic digester that helps in the degradation of basic components of organic material during anaerobic digestion of brewery wastewater.
Due to these facts, sequencing of 16S rRNA gene [8, 22] with high-throughput metagenomic technologies such as 454 pyrosequencing and Illumina (e.g., HiSeg, MiSeq) techniques has been developed and successfully adopted for the identification of diverse microbial communities in Anaerobic Digestion (AD) system [23-26]. These techniques are found suitable to investigate the microbial ecology in the anaerobic system given the fact that most of the organisms are not culturable [27]. High resolution power of Next Generation Sequencing (NGS) makes it possible to detect some population that is very small with rare species and possibly potential functionalities in anaerobic granules. The identification of diverse groups in the anaerobic system could serve as a link between microorganisms and physical properties of the granules and thus provide an opportunity for granules modification, improve granule strength as well as shorten reactor start up time and stability. It is important to note that, studies of the microbial community in anaerobic granules using NGS are still limited especially for anaerobic reactor treating industrial wastes such as brewery wastewater.
Nonetheless, the aim of this study is to investigate the characteristics, abundance and phylogenetic diversity of the microbial community in the granular sludge obtained from a full-scale biogas UASB reactor treating brewery wastewater. To this end, we have succeeded in identifying organisms which are potentially related to reactor operations in relation to biogas generation. Currently, there exist few reports on the application of next-generation sequencing (NGS) such as MiSeq Illumina sequencing platform to determine the complexity of the microbial communities present in UASB reactor treating brewery wastewater. In view of this, this study has the potential of contributing to the microbial biodiversity database that is presently available on bacteria inhabiting the UASB reactor treating brewery wastewater. It will facilitate the development of more efficient reactor performance to reduce organic matter and produce more methane that could serve as a source of renewable energy.
2. MATERIALS AND METHODS
2.1. Sample Collection and Physico-chemical Analysis
Methanogenic sludge samples used in the present study for shotgun sequencing were obtained from a full-scale industrial UASB reactor treating brewery wastewater in Durban, South Africa, as earlier described by Enitan et al. (2014). One liter of homogenized granular sludge samples was obtained for microbial analysis while influent and effluent from the reactor were collected in one-liter sterile glass bottles each for physico-chemical analyses as earlier described in detail by Enitan et al. [15]. All the samples were transported to the laboratory at 4°C before analysis. Granular samples were further separated into two: The first one was fixed in 2.5% glutaraldehyde overnight for scanning electron microscope, while the other was used for DNA extraction.
2.2. Scanning Electron Microscope
Scanning Electron Microscope (SEM) was carried out in a Zeiss Ultra Plus Field Emission Gun Scanning Electron Microscope (FEGSEM). Prior to SEM analysis, the granules were washed with phosphate buffer and fixed with 2.5% glutaraldehyde overnight at 4˚C. Fixed granules were washed with PBS buffer, dehydrated by successive passages through 30, 50, 75 and 100% alcohol. The aggregates were then mounted on a stub with double-sided tape and dried with a critical point dryer. The granular samples were observed at an accelerated voltage of 20 kV in a Jeol JSM-5910LV after being coated with gold sputter coated (Electron Microscopy Unit, University of KwaZulu-Natal, Durban, KwaZulu-Natal, South Africa).
2.3. Genomic DNA Extraction, DNA Library Construction and Sequencing
Genomic DNA was extracted within 24 hours after sampling in duplicate. Samples were homogenised and two millilitres of sample was centrifuged at 9,600 x g for 5 minutes to release the microorganisms entrapped within the granules and other undigested particles. The pellets were washed twice with 1 x PBS and centrifuged again at 9,600 x g for 5 minutes to collect the pellets. Total genomic DNA was extracted from the pellets using Fast DNA SPIN Kit following manufacturer’s instructions (MP Biomedicals, LLC, Solon, OH). Finally, the DNA was eluted in 50μL of TE buffer and the quality was assessed on 1% agarose gel. Purity and yield of extracted genomic DNA were checked by Nanodrop Spectrophotometer (ND-1000) and Qubit fluorometer. The extracted genomic DNA was stored at -20°C for 16S metagenomic sequencing using Illumina MiSeq paired-end sample preparation kits according to the manufacturer instructions. The DNA sample was amplified using barcoded bacterial primer pair S-D- Bact-0341-b-S-17/S-D-Bact-0785-a-A-21 (targeting the V3 - V4 hypervariable region) with an amplicon size of 464 bp. The PCR was done based on the method reported by Klindworth et al. (2013).
2.4. Taxonomic Annotation of Metagenomes
Metagenomic sequences were generated online through the metagenome analysis tool MG-RAST version 3.0 (http:// metagenomics. anl. gov/ ). Before analysis, the read paired ends were merged according to the instructions provided by MG-RAST. The irrelevant and artificial replicate sequences were automatically removed and low-quality sequences were filtered out using default settings on MG-RAST. The read sets were normalized by random subsampling and rarefaction curves constructed to assess whether sequencing effort was sufficient to capture the majority of taxonomic diversity.
Reads generated were taxonomically annotated blasted on M5 Non-Redundant (M5NR) protein database using sBLAT. M5NR database integrates many sequence databases such as European Bioinformatics Institute (EBI), Gene Ontology (GO), Joint Genome Institute (JGI), Phage Annotation Tools and Methods (Phantome), National Center for Biotechnology Information (NCBI), The SEED Project (SEED), Kyoto Encyclopedia of Genes and Genomes (KEGG), UniProt Knowledgebase (UniProt), Virginia Bioinformatics Institute (VBI), evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG) into one single, searchable database on the MG-RAST server. Analysis of sequences that passed the QC after filtering was done to construct circular tree using MG-RAST workbench.
3. RESULTS AND DISCUSSION
3.1. Reactor Performance and Structure of Granular Sludge Using SEM
The physico-chemical parameters of the influent and effluent were used to determine the performance and stability of the UASB reactor. It was further used to evaluate the operational conditions and the ability of the reactor to support the growth of microorganisms that are needed for the conversion of organic matter to biogas as well as good effluent quality. In this study, an average Chemical Oxygen Demand (COD) concentration recorded was 8295.71 mg/L with an average COD removal efficiency of 80% at mesophilic temperature (28-32˚C) and 12 h Hydraulic Retention Time (HRT). The optimal pH values recorded under various organic loading rates ranged between 6.5 and 7.4 with 70% of methane production in the biogas generated.
Morphology and the size of the studied UASB granules were determined using SEM in order to evaluate the extent at which the reactor supported granulation and microbial growth. The SEM micrograph reflects that the operational condition of the reactor supports the Extracellular Polymeric Substances (EPS) in the granule formation. The granules have an average diameter of about 1.24 mm. SEM analysis showed an oval shape and clear surface morphology with an irregular projection on the surface of the granules (Fig. 1). Therefore, the morphology and characterization of granule formation in this study would help reactor operators to determine the optimum condition that the reactor must be operated during treatment of brewery wastewater with the aim of getting a better floc structure and prevention of biomass washout from the reactor.

According to Yetilmezsoy et al. [28] and Ahn’s proposed model [29] for anaerobic sludge granulation, SEM micrographs appear to represent the growth of a small granule in the diameter range of 1-2 mm. Similar studies on the morphological study of the granules in UASB and hybrid reactors by Gupta et al. [30, 31] and Chatterjee et al. [32] elaborate more on the granulation ability of a reactor to influence the structure and retained the diverse communities of microorganisms within the reactor [31]. Extracellular polymeric substances in granular sludge are also a major contributor to sludge floating and efficiency of any reactor, therefore the determination of the size and structure of the granules in this reactor is very important. Other advantages of good anaerobic sludge granulation in UASB reactors are reviewed by Li [33] and Hulshoff et al. [13].
3.2. Assessment of Microbial Community Structure
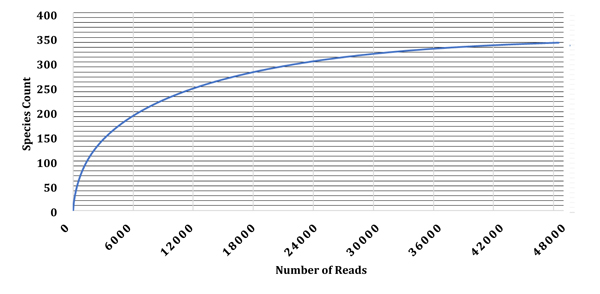
Characterization of microbial community structure in the granules collected from the full-scale UASB reactor was performed through high-throughput sequencing (MiSeq) of 16S rRNA gene. The sequence reads from this study were deposited in MG-RAST (Accession number 4713539.3) with overall raw paired-end reads of 48,488 sequences and total basepairs of 14,602,358. About 6,189 pre-filtered sequence reads (12.8%) failed quality control pipeline; 39,356 sequences (81.2%) that passed quality control had ribosomal RNA genes while about 2,943 (6.1%) of the sequences had no rRNA genes. The obtained itags were aligned and subjected to Blastn against known 16-18S rRNA gene tag M5NR database. The rarefaction curves plateau to the right at maximum E-value cutoff of 1e-5 based on the available source databases used for analysis (Fig. 2). The rarefaction curve showed the species richness in the granules collected from the UASB reactor. Alpha diversity of this metagenome summarized the diversity of organisms in a sample with a single number with α-diversity of 24.186 species.
The phylogenetic characterization of the microbial community structure and composition from the total genomic DNA sequence reads in Fig. (3) revealed the dominance of bacterial community (94.4%) in the reactor followed by Archaea community (1.8%), Viruses (0.7%), Eukaryota (0.9%), and unknown organisms (2.2%). The taxonomy affiliation of the bacterial community at different structural level was analyzed for better understanding. In total, 18 phyla were identified apart from the unclassified sequences (Fig. 3). The sequence results revealed phylum Firmicutes (32.26%) as the dominant community followed by the phyla Bacteroidetes (27.78%), Synergistetes (5.38%) and Proteobacteria (4.29%) while, the unclassified bacteria accounted for 26.79%. This is in accordance with other reported metagenomics studies for biogas-producing microbial communities [20, 34]. A similar result on the dominant of phyla belonging to Proteobacteria, Bacteroidetes, Spirochaetes, Clostridia, Chloroflexi and Synergistia were identified in the granular sludge samples collected from nine full-scale methanogenic bioreactors treating brewery wastewater using 454 Pyrosequencing [35]. Other studies on the abundance of these phyla in the recovered 16S rRNA gene sequence reads in anaerobic digesters fed with different feed stocks using NGS have also been reported [24-26, 36].
Classification into lower taxonomic levels showed diverse population in the reactor samples with up to 29 orders, 36 families and 44 genera (Fig. 3) as the dominant group in the granular sludge samples. The taxonomic distribution revealed that the orders Flavobacteriales (24.47%), Synergistales (6.44%), Clostridiales (3.70%) and Bacteroidales (2.63%) were the predominant order among the 16S rRNA encoding reads. The key bacteria that are involved in hydrolysis, acidogenesis, acetogenesis and methanogenesis processes during AD processes are present in the UASB reactor (Figs. 3 and 4). The rank abundance plots representing the taxonomic richness and abundance of unclassified sequences at species level is shown in Fig. (4).
The hydrolytic bacteria from the genera Streptococcus and Enterobacterium such as E.coli, Klebsiella that help in the conversion of insoluble organic compounds to soluble monomers and dimers [37] have been found in abundance in this reactor. Bacteroidetes that perform the acidogenic process are well represented in the reactor as per the sequencing results (Fig. 3). They are bacteria that metabolise carbohydrates and peptone products to acetate, formate, lactate, or propionate. Hence, the result of this study is in accord with previous studies [24, 37]. Similarly, subgroups of Bacteroidetes belonging to families Porphyromonadaceae and Rikenellaceae were dominants in the investigated biogas plants (Fig. 3) [34]. These microorganisms are involved in volatile fatty acid (VFA) production as well as proteins and carbohydrates degradation, thus, their presence will help in maintaining and keeping the level of VFA production and consumption in the reactor [20, 26].
Members of delta, alpha and gammaproteobacteria and the low G+C Gram positive classes that help in converting sugars, fatty acids and amino acids to organic acids (formic, acetic, propionic, butyric, lactic acids), ketones and alcohols are well represented in the reactor (Fig. 3). Sequence similarity was also closely related to Firmicutes as the dominant phylum belonging to families Clostridiaceae and Leuconostocaceae. This group is known to be directly involved in the conversion of complex organic matters in the industrial waste to metabolites that could be used directly by the methanogenic Archaea. These organisms are efficient in the degradation of complex organic matter and acetic or lactic acid fermentation to CO2 and H2 [5, 25, 38, 39]. Genus Clostridium of the family Clostridiaceae is known as hydrogen-producing organism while facultative anaerobes belonging to genus Leuconostoc that depend on amino acids and fermentable carbohydrates for their growth are present in abundance in the reactor [38].

Other diverse group of class Deltaproteobacteria (formally known as Deltaproteobacteria group TA) belongs to the family Syntrophorhabdaceae that contains wellknown species of syntrophic substrate-degrading anaerobes, such as the genera Syntrophus and Syntrophobacter detected by MiSeq Illumina sequencing in this study [40, 41]. These organisms are known as amino acids degraders and sulphate-reducing bacteria. Species of the genus Syntrophobacter have the ability to utilize sulphate as an external electron acceptor, but their growth by sulphate reduction is known to be very slow [41, 42].
Other detected SRB belongs to general Desulfovibrio, Desulfutomaculum, Syntrophomonas and Syntrophobacter. They are acetogenic groups that form a synergistic cooperation with methanogenic Archaea during anaerobic digestion systems (Figs. 4 and 5) [43, 44]. These organisms are involved in incomplete oxidation of molecular hydrogen and organic compounds (e.g. butyrate, propionate, ethanol, lactate, etc.) to acetate and CO2 during anaerobic fermentation [37, 45]. Desulfovibrio sulfuricans and D.alaskensis are abundant in the reactor (Fig. 5), where they produce acetate that could be consumed by methanogens especially genus Methanosarcina (Figs. 4 and 5). The identified Desulfovibrio species have been reported to produce acetate, CO2 and H2 in co-occurrence with hydrogenotrophic methanogens during limited sulfate conditions.
Competition and coexistence of Sulphate-Reducing Bacteria (SRB) acetogens and methanogens in an anaerobic bioreactor were earlier investigated by Dar et al. [46]. Researchers have also reported the syntrophic relationship between hydrogen producing acetogens such as Syntrophobacter and hydrogenotrophic methanogens such as Methanothermobacter and Methanosarcina [43]. In addition to the distribution of phylogenetic properties, Shewanella, Gram-negative bacteria that can reduce nitrate to nitrite as well as reduce trimethylamine N-oxide and sulphur to produce hydrogen sulphate from thiosulphate were also detected in this reactor.
Apart from bacterial communities, sequences assigned to methanogenic Archaea belonging to the order Methanosarcinales were also identified with other sequences or hits that are assigned to the unclassified archaeon in the metagenomes (Fig. 4). This is similar to our previous studies using amplicon sequencing of archaeal rRNA gene and florescent in-situ hybridization [45, 47], where acetoclastic Methanosaeta belonging to the order Methanosarcinales was found in the reactor. Methanosarcinales are seen as methanogenic archaea that mediate both aceticlastic methanogenesis and hydrogenotrophic methanogenesis phases depending on the reactor condition [26, 48] especially at mesophilic condition [49]. Relatively, a large number of these types of bacteria isolated mainly from methanogenic environments especially UASB sludge samples have been reported in the literature [7, 43, 50].
The 16S rRNA gene sequence reads in this study show diverse bacteria and methanogenic Archaea population even at lower to no sulphate concentration in the reactor. This highlights a possible syntrophic association of great importance for enhancing biogas production. The taxonomic distribution showed that some of the sequences could not be assigned to species level while variation in the methanogenic archaea in the reactor could be linked to operating parameters such as reactor pH, retention time, temperature [48, 50], substrate and other parameters as listed by Cai [26]. Therefore, optimizing reactor conditions could be a practical strategy for enhancing bacterial communities in the reactor, increase their potential functions, thus enhancing the yield of biogas production.


CONCLUSION
In conclusion, the major bacterial communities detected in the UASB reactor investigated using the V3 - V4 region of 16S rRNA gene on the Illumina MiSeq high-throughput sequencing platform are known organisms that are essential for the degradation of organic matter in anaerobic reactor for biogas production. Most of the organisms identified are major producers of metabolites that are used by methane-producing archaea. The study showed a diverse group of bacteria (Figs. 3 and 5) that are present in the UASB reactor, however, substantial percentages of the sequence reads could not be assigned to any group, thus remaining unknown. We found that metagenome of sludge samples was similar to previous reports on the microbial communities in biogas producing industrial wastewater treatment system. However, the result suggested that further studies could be explored on the metaproteomic and metatranscriptomic analyses in order to identify the unknown populations as well as determining their role in organic degradation and metabolic functions during anaerobic digestion of brewery wastewater.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The authors confirm that the data supporting the findings of this research are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Due credit is given to Durban University of Technology against the background that this article is largely a spin-off of a Ph.D thesis that was graciously supported by its Administration. The University of Venda, Limpopo, South Africa, equally deserves worthwhile commendation for their accepted offer of financial assistance in covering the costs of publishing this article in an open access journal.

