All published articles of this journal are available on ScienceDirect.

Molecular Diagnostic Methods for Detection and Characterization of Human Noroviruses
Abstract
Human noroviruses are a group of viral agents that afflict people of all age groups. The viruses are now recognized as the most common causative agent of nonbacterial acute gastroenteritis and foodborne viral illness worldwide. However, they have been considered to play insignificant roles in the disease burden of acute gastroenteritis for the past decades until the recent advent of new and more sensitive molecular diagnostic methods. The availability and application of the molecular diagnostic methods have led to enhanced detection of noroviruses in clinical, food and environmental samples, significantly increasing the recognition of noroviruses as an etiologic agent of epidemic and sporadic acute gastroenteritis. This article aims to summarize recent efforts made for the development of molecular methods for the detection and characterization of human noroviruses.
1. INTRODUCTION
Human noroviruses are a group of highly related viruses that were first identified from fecal specimens obtained in 1968 during the investigation of an outbreak of acute gastroenteritis in Norwalk, Ohio. The prototype strain designated as Norwalk virus was discovered by Kapikian and colleagues in 1972 [1]. Viruslike particles of 27-nm in size were visualized from Norwalk-derived patient’s fecal filtrate using electron microscopy (EM). The viruses are now recognized as the most common cause of epidemics of acute gastroenteritis and an important cause of sporadic gastroenteritis in both children and adults [2]. The incidence of norovirus illness is high. Annually, human noroviruses account for over 267 million infections worldwide [3]. In developing countries, it is estimated that over 1 million hospitalizations and 220,000 deaths occur among children less than 5 years old each year [4]. In some industrial countries including United States where childhood immunization has substantially reduced the incidence of rotavirus-associated gastroenteritis [5], noroviruses become the leading causative agent of acute pediatric gastroenteritis [6]. A major impediment to the diagnosis of norovirus infection is the absence of an effective cell culture system or small animal models for propagating the viruses. Owning to the exceptionally low infectious dose, a few virus particles can initiate a norovirus infection. Conventional detection methods e.g. electron microscopy and immunoassays apparently lack sufficient sensitivity. In the absence of simple and sensitive detection approaches, the etiologic role of noroviruses in the sporadic and outbreak-associated acute gastroenteritis has been immensely underestimated. Conventional diagnostic methods left 62% to 94% of cases in the diagnostic void [7-8]. This diagnostic gap has not been reduced until new and more sensitive molecular detection methods have been introduced in the field since the1990s. Since then, a wide variety of molecular methods have been developed and exploited for the detection of noroviruses in clinical, food and environmental samples. This review aims to summarize recent efforts made for the development of molecular methods for the detection and characterization of noroviruses, an agent of acute viral gastroenteritis.
2. GENOME ORGANIZATION AND CLASSIFICATION
Noroviruses are a group of highly related and extremely infectious, small round-structured, nonenverloped viruses with a positive-sense single-stranded RNA genome of approximately 7.6 kb in length. The viral RNA is covalently bonded to the viral protein genome (VPg) at the 5’end and contains a polyadenlated -A tail at the 3’end. The genomic viral RNA codes for three open reading frames (ORFs), designated ORF1, ORF2, and ORF3 (Fig. 1). ORF1 is translated as a 194-kDa polyprotein that is cleaved by the virus cysteine protease into 6 nonstructural proteins including p48, nucleoside triphosphatase (NTPase), p22, VPg, Protease (Pro) and RNA-dependent RNA polymerase (Pol). ORF2 encodes a 60-kDa major viral capsid protein (VP1), a structural protein involving virus replication. ORF3 encodes a 23-kDa structural minor capsid protein (VP2) interacting with the viral RNA when the virion formation occurs [9].
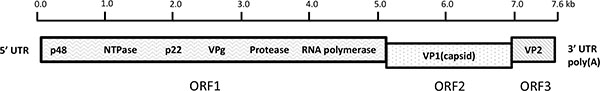
Schematic of human norovirus genome organization. The viral genome is composed of a single-stranded, positive-sense RNA, ~7.6 kb in length and is divided into three open reading frames (ORFs) designated ORF1, ORF2, and ORF3. ORF1 encodes 6 nonstructural (NS) proteins including an NTPase, 3C-like protease, and RNA-dependent RNA polymerase (Pol). ORF2 and ORF3 encode major structural capsid protein 1 (VP1) and minor capsid protein 2 (VP2), respectively. Nucleotide position numbering refers to the Norwalk virus genomic sequence (GenBank accession no. NC001959).
Noroviruses belong to highly antigenically and genetically diverse genus Norovirus in the family Caliciviridae, which also includes Sapovirus, Lagovirus, and Vesivurs as well as Nebovirus [10]. Historically, antigenic classification of noroviruses was determined on the basis of cross-challenge studies in volunteers and cross reactivity analysis by solid-phase IEM [1, 11-13]. These studies demonstrated that considerable antigenic diversity among norovirus strains has been observed. However, due to the cross-reactivity of the antibodies, the antigenic classification schemes displayed limited accuracy and reproducibility [14]. It is thought that these limitations were mainly linked to the cross-reactive antibody responses observed in individuals [15, 16].
Because of the absence of in vitro cell culture system for the replication of human noroviruses, direct serotyping of the viruses using a conventional neutralization test has been severely hampered [17]. Genetic classification of the viruses was defined only from 1990 after the successful cloning of the Norwalk virus genome [18]. Since then, genomic sequencing of the amplicon derived from reverse transcription polymerase chain reaction (RT-PCR) and subsequent phylogenetic analysis have become the principle means for characterizing the viruses and understanding the genetic relatedness of virus strains [18, 19, 14]. Based on the complete sequence of capsid protein, noroviruses are genetically classified into at least 5 genogroups (GI-GV) [20], with viruses within genogroup I, II, and IV associated with human gastroenteritis. Genogroup III and V have been identified in cows and mice, respectively. Virus variants consisting of canine norovirus were recently discovered [21, 22]. GI and GII contain the majority of norovirus strains and present the greatest genetic diversity. They can be further classified into around 31 genotypes, with 9 genotypes in GI and 22 in GII, respectively [20, 23]. Prototype Norwalk virus was assigned as genogroup I, genotype 1 (GI.1). The presence of the extensive variability of norovirus strains is resulted from both accumulation of point mutations related to error-prone RNA replication and recombination between related viruses [24, 25]. Of all the human norovirus strains, GII.4 has become the predominant strains causing the most outbreaks and sporadic gastrointestinal disease in the United States and worldwide despite the great diversity of noroviruses [2, 26].
3. TRANSMISSION OF NOROVIRUSES AND THEIR GENOTYPES
Noroviruses are recognized the cause of person-to-person transmission of acute gastroenteritis in a broad breadth of healthcare settings e.g. hospitals, nursing homes, and community settings e.g. schools, day-care centers, cruise ships, and restaurants. Transmission most often occurs via the fecal-oral route, by person-to-person contact, by consuming foods contaminated at the source through environmental contamination or prepared by infected food handlers, by exposure to aerosolization of the virus and subsequent contamination of surfaces. Of all, person-to-person contact is the most common route, accounting for 83.7% of all reported outbreaks in the United States from 2009 to 2013 while foodborne transmission was 16.1% [27]. GII.4 was considered to be more frequently associated with person-to-person transmission while GII.12 and GI.7 were likely associated with foodborne illness [27]. Globally, approximately 14% of all norovirus outbreaks are attributed to food with 37% of outbreaks caused by mixtures of GII.4 and other viruses, 10% by GII.4, and 27% by all other single genotypes [28].
4. MOLECULAR DIAGNOSTIC TESTS
Electron microscopy (EM) was initially utilized for direct detecting norovirus particles with 27 nm in diameter from fecal specimens. However, it is not a high throughput assay that can be practically deployed in epidemiological or clinical studies because of insufficiently distinctive surface morphology of the viruses, and low assay sensitivity (106-7virus particles per gram stool) as well as requirement for costly instrument and well trained microscopist [29]. Immunoassay-based detection represents a substantial advance for norovirus diagnostics. However, norovirus immunoassay testing has demonstrated poor sensitivity when applied on clinical specimens [30]. Due to the lack of rapid and sensitive tests, considerable amounts of outbreaks of nonbacterial gastroenteritis went undected and were mistakenly thought to have unknown etiologic agent. Noroviruses have been considered to play an insignificant role in sporadic and outbreak-associated acute gastroenteritis [7, 31]. The successful cloning and sequencing of Norwalk and Southampton agents led to breakthrough in understanding the molecular biology of noroviruses and paved the path to the development of more molecular-oriented detection methods [18, 32]. The availability and application of more sensitive molecular approaches has rapidly changed our understanding of norovirus disease. Fig. (2) depicts some of the noteworthy changes in the detection of human noroviruses since 1972. The introduction of RT-PCR in 1992 marked the beginning of molecular diagnostics of noroviruses [33].
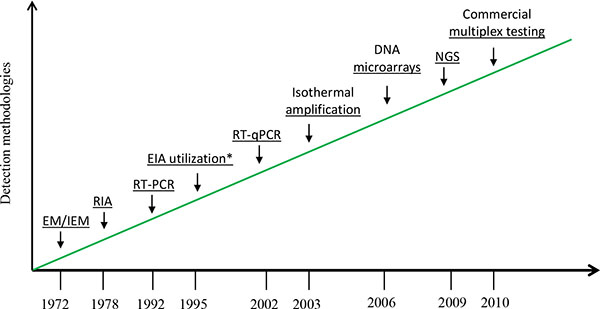
Advances in detection of human noroviruses since 1972.
EM/IEM: electron microscopy/immune EM, RIA: radioimmunoassay, EIA: enzyme immunoassay, RT-qPCR: real-time quantitative reverse transcription polymerase chain reaction, NGS: next-generation sequencing
*EIA for antigen detection
4.1. Detection - Monoplex Reverse Transcription RT-PCR and Isothermal Amplification
4.1.1. Standard RT-PCR
Viral RNA extracted from clinical samples e.g. stools, vomitus and serum is transcribed to complementary DNA (cDNA) in the presence of reverse transcriptase using either random primers or norovirus specific primers. The resulting cDNA then can be amplified by DNA polymerase using norovirus specific primers. Primer design plays a critical role in the sensitivity and specificity of RT-PCR assays. The selection of inappropriate primers may result in false-negative consequence, making the diagnosis void. Given the genetic diversity of noroviruses, regions within the viral genome with the highest degree of conservation among strains are targeted for primer selection for RT-PCR amplification. Early conventional RT-PCR assays utilized a number of primers mainly selected from the highly conserved RNA polymerase region in ORF1of the prototype Norwalk virus genome [33-37]. Many of the assays employing the primers resulted in an unexpectedly low detection rate of EM-positive clinical specimens owing to the wild genetic diversity among virus strains. In order to resolve this diagnostic problem, a number of efforts have been made to design broadly reactive primers for the RT-PCR assays with the availability of accumulating sequence information of noroviruses strains [38-41]. The new design of the primers in the studies was still primarily based on the RNA polymerase region, but they significantly improved the assay sensitivity and specificity via southern hybridizations and DNA sequencing.
Although the polymerase region has been favored to amplify noroviruses, the great genetic diversity of the viruses has still made it difficult to detect all the strains using the primers chosen from the region. It was reported that viruses within the same genogroup displayed only 60.8% - 63.3% nucleotide and 67.3% - 69.1% amino acid identity [42]. To attain additional sequence information that might be useful in identifying other unique strains, primers designated to amplify other regions of the viral genome e.g. capsid protein region have been evaluated [34, 43-49]. The capsid region sequence represents the most variable part of the viral genome. Variation rates among strains of the same genogroup range from 45% to 61% and within genotypes at a rate of 14 - 44% [20]. In general, highly conserved RNA polymerase sequences are more broadly reactive for norovirus detection than capsid sequences due to lower genetic diversity in that region, but such sequences become problematic for phylogenic classification that cannot correctly separate some strains into proper clusters due to limited sequence variations [47].
4.1.2. Nested or Semi-Nested RT-PCR
Nested or semi-nested RT-PCR assays have also been developed to increase the sensitivity and specificity for the detection of norovirus strains in GI and GII [43, 50-55]. This approach used two successive runs of PCR employing one (semi-nested) or both (nested) primers in the second round amplification targeting the RNA polymerase or capsid region. This strategy has been reported to achieve at least as sensitive as RT-PCR when used in combination with southern hybridization [54], or 10 to 1000-time more sensitive than single round RT-PCR [51].
4.1.3. Isothermal Amplification
Isothermal amplification is sensitive and convenient in amplifying DNA targets, and can be integrated with a RT step for RNA viruses. Isothermal amplification methods such as nucleic acid sequence-based amplification (NASBA) [56-60] and RT loop-mediated isothermal amplification (RT-LAMP) [61-63] have been developed for norovirus detection. NASBA attained similar analytical detection limits to that of real-time TaqMan RT-PCR assay but provided less consistent signals [59]. Compared to routine RT-PCR, the clinical sensitivity and specificity of the RT-LAMP assay were 100 and 94% for GI as well as 100 and 100% for GII, respectively, by analyzing a total of 91 fecal specimens [61].
4.2. Quantification/Monitoring - RT-qPCR
Compared to standard RT-PCR in which the product of the reaction is detected at its endpoint, the key feature of real-time quantitative RT-PCR (RT-qPCR) is that the amplicon is detected by continuous measurement of the PCR product as the reaction progresses. This feature eliminates the need for post-amplification processing e.g. agarose gel visualization. Moreover, quantitative determination of target nucleic acid is possible in RT-qPCR assays, although it is not generally used in diagnostics. RT-qPCR commonly utilizes two methods for the detection of noroviruses: 1) non-specific fluorescent dyes e.g., SYBR green that intercalate with any double-stranded DNA [64-67]; and 2) sequence-specific oligonucleotide probes including TaqMan and hybridization probes that are labelled with a fluorescent reporter that enables detection only after specific binding of the probe with its complementary sequence [68-79]. SYBR green assay is less costly and simpler than TaqMan assay in its design and manipulation. Direct comparison between SYBR green and TaqMan assay on the detection of noroviruses has not been evaluated. No significant difference between them in terms of specificity, sensitivity and quantitativity in the detection of other pathogen was observed [80]. While the relative conserved viral polymerase region was still favored as the amplification target in many real-time qPCR assays, a GI and GII ORF1-ORF2 junction region, which has been shown the highest sequence similarity across the viral genome, was firstly targeted for the design of primers and TaqMan probes in a norovirus detection assay described by Kageyama and colleagues [68]. Their assay showed a better detection rate in 80 (99%) of 81 stool samples than conventional RT-PCR assays that detected 77% for the polymerase and 83% for the capsid N/S region, respectively, in the same panel of stool specimens. In addition to two-step assays, RT-qPCR also can be performed in one-step format for simultaneous detection and genotyping of noroviruses [69, 70, 75]. One-step RT-qPCR that combines reverse transcription and cDNA amplification into a single reaction decreases the risk of carryover contamination and experimental variation since both reactions take place in the same tube, but its following disadvantages are evident: difficult to optimize the two reactions separately; detection of fewer targets per sample; and likely less sensitive than two-step assay since the reaction conditions are a compromise between the two combined reactions. Overall, with improved specificity and sensitivity, and capability of viral load quantitation, RT-qPCR has now been widely used as a reference laboratory approach for detecting norovirus in fecal specimens, water and food [2]. However, its multiplex application which has been limited by the availability of fluorescent reporter dyes is a remained issue.
4.3. Differentiation/Genotyping - Multiplex Testing, DNA Microarrays, and NGS
4.3.1. Multiplex Testing
Noroviruses, along with other gastrointestinal pathogens, can be detected using multiplex molecular diagnostic technologies. Multiplex molecular technologies offer rapid and simultaneous detection of multiple nucleic acids in high throughput format within a single reaction, greatly cutting down time, labor and cost associated with single reaction detection technologies. Recently, several multiplex nucleic acid approaches such as BioFire FilmArray Gastrointestinal (GI) Panel (Salt Lake City, UT), Luminex xTAG Gastrointestinal Pathogen Panel (GPP) (Toronto, Canada) and Nanosphere Verigene Enteric Pathogens (EP) Test (Northbrook, IL) for the detection of pathogenic enteric pathogens including noroviruses received clearance from U.S. Food and Drug Administration (Table 1). FilmArray GI Panel is designed to detect 22 bacterial, parasitic and viral agents, including norovirus GI/GII. This device can extract and purify all nucleic acids from the samples, and performs a single, large volume, massively multiplex (RT) PCR reaction followed by individual singleplex second-round PCR reactions to detect the first round amplified products. Buss [81] reported 94.5% (52/55) clinical sensitivity and 98.8% (1483/1501) specificity of norovirus detection in the performance evaluation of FilmArray GI Panel by testing a total of 1,556 stool specimens comparing to the results obatained by polymerase-capsid junction PCR. Khare [82] also demonstrated high (> 90%) sensitivity and specificity for norovirus GI/GII in their study using FilmArray GI Panel. In contrast to FilmArray GI Panel, xTAG GPP requires separate extraction and purification of sample nucleic acid prior to amplification and detection. This approach utilizes multiplex RT-PCR using target-specific TAGed primers and biotinylated primers followed by bead hybridization and detection. xTAG GPP is capable to simultaneously detect 3 viral agents including norovirus GI/GII, 9 bacterial, and 3 parasitic pathogens responsible for infectious diarrhea. Clinical performance characteristics of norovirus GI/GII in the xTAG GPP assay has been evaluated in several studies [83-87]. In these evaluation studies, it demonstrated that the multiplex nucleic acid assays offer an advantage to identify high percentage of mixed viral-bacterial infections in the clinical specimens. Noroviruses were among the most frequently detected pathogens in mixed infections [81, 82, 87]. As the clinical implication of the viruses and bacteria involved in mixed gastroenteritis infections is not well understood, it creates a challenge for the data interpretation to identify the etiologic role of noroviruses during the course of the gastrointestinal disease.
Commercial multiplex testing to detect human noroviruses along with other enteric pathogens.
| Company | Platform | Method | No.of Pathogens Targeted | Turnaround Time (h) | FDA Cleared |
|---|---|---|---|---|---|
| Biofire Diagnostics | FilmArray GI Panel | Nested RT-PCR | 22 | 1-2 | + |
| Luminex | xTAG GPP | RT-PCR + Bead hybridization | 15 | ~5 | + |
| Nanosphere | Verigene EP | RT-PCR + nanoparticle hybridization | 7 | ~2 | + |
| Seegene | Seeplex Diarrhea-V ACE | RT-PCR + capillary electrophoresis | 5 | ~10 | - |
| Fast-Track Diagnostics | FTD Viral GE | Real-time RT-PCR | 5 | ~6 | - |
Verigene EP Test is an automated, multiplexed molecular diagnostics that uses PCR followed by hybridization to gold nanoparticles to detect specific gastrointestinal microbial nucleic acid genes associated with 7 pathogenic bacteria and viruses including Campylobacter group, Samonella sepcies, Shigella species, Vibrio group, Yersinia enterocolitica, Norovirus GI/GII and Rotavirus A. Of the three multiplex devices, FilmArray and EP Test are sample-to-result systems offering 1-2 h turnaround time, with minimal hands-on time, while xTAG GPP requires ~ 5 h with exclusion of sample processing. Despite the longer turnaround time, xTAG GPP can analyze up to 96 samples in an 8-h shift while FilmArray can only process 1 sample at a time. Vitally, xTAG GPP is an open system in which the amplicons need to be handled, there poses an increased likelihood of cross contamination.
In addition to the three FDA-cleared multiplex approaches, Seeplex Diarrhea-V ACE (Seegene, South Korea) and FTD viral GE (Fast-Track Diagnostics, Luxembourg) are other two commercial systems to detect multiple enteric viruses. Seeplex Diarrhea-V ACE is a multiplex RT-PCR system that detects 5 diarrheal pathogens including noroviruses GI/GII [88]. FTD viral GE is a two-tube test system using multiplex real-time PCR for detection of norovirus GI, GII, astrovirus, rotavirus, adenovirus and sapovirus. Liu and colleagues [89] developed a TaqMan Array Card-based method (Life Technologies, Grand Island, NY), a 384-well singleplex real-time PCR format, for simultaneous detection of 19 enteropathogens. The TaqMan Array card showed 100% (31/31) clinical sensitivity and 96.2% (75/78) specificity for norovirus GII, compared to laboratory-developed PCR-Luminex assay.
Luminex xMAP is an alternative multiplex PCR method for detection of multiple viral pathogens. Liu [90] developed a Luminex xMAP-based assay for simultaneous detection of seven enteric viruses associated with acute gastroenteritis, yielding 86.67% sensitivity and 100% specificity in the detection of noroviruses (GI/GII) from clinical fecal specimens when compared with RT-PCR. The study by Hamza [91] revealed the multiplex xMAP assay could simultaneously detect multiple human enteric viruses including noroviruses from environmental water samples.
4.3.2. DNA Microarrays
DNA microarrays have been used for simultaneous detection and genotyping noroviruses as well as other enteric viruses in previous studies [92-96]. In these reports, RT-PCR with virus-specific primers has been used to amplify the target nucleic acids prior to microarray hybridization, making them still limited in use due to the substantial genetic diversity of the viruses. We recently reported a novel sequence-independent linear RNA amplification of minute amounts of RNA for microarray-based simultaneous detection and identification of multiple enteric viruses including a GII norovirus [97]. This method offered increased microarray detection accuracy over conventional T7 in vitro transcription amplification. Further evaluation of this system in a small-scale study indicated that various genotypes of noroviruses (GI/GII) were detected in clinical specimens, showing 100% concordance at genogroup level and 85% at genotype level with regard to RT-PCR and DNA sequencing [98]. In addition to discerning genotypic information on noroviruses, this platform allowed the detection of co-infection with more than one viral pathogens present in the same sample. The use of this platform does not require PCR amplification and may circumvent the primer selection problem resulting from PCR steps owning to the great genetic diversity of the viruses; yet provides a sufficient level of sensitivity. Microarray detection of random PCR amplified nucleic acids from a range of gastrointestinal viruses including Norwalk virus was recently described [99]. DNA microarrays have proven an important means for discovery in numerous studies. But, given the limitations such as cost, sensitivity, validation, and data interpretation/ management, DNA microarrays are not currently implemented in clinical microbiology laboratories or epidemiological investigations for the study of norovirus infections /outbreaks.
4.3.3. Next-Generation Sequencing (NGS)
Large amounts of sequence diversity have been observed across ORF2 in norovirus genome. For reliable genotyping of the virus strains, a sequence determination of the complete major capsid region is recommended [20]. More recently, a dual-nomenclature scheme using both ORF1 and ORF2 sequences has been proposed [23] as inter-genotype recombination in the ORF1/ORF2 is common. However, partial nucleotide sequences of ORF1 or ORF2 of the virus genome are also used for genotyping the viruses since sequencing of the full major capsid region is not a routine practice with traditional sequencing approaches. Recent development of high-throughput next-generation sequencing (NGS) technology has the potential to revolutionize our ability to nucleic acid sequencing, providing an unprecedented way to rapidly and deeply sequence the genomes of known or unknown microbial organisms. Utilization of NGS technology for the discovery and characterization of noroviruses have been described in a number of studies [100-103]. In addition to virus detection and typing in epidemiology investigations, NGS has been applied for norovirus evolution studies to characterize viral communities and dynamics [104, 105]. NGS has opened new avenues for virus research and diagnostic applications. It is envisaged that the development of NGS in will bring new and more understandings of the molecular virology of norovirus that leads to major diagnosis advances.
CONCLUSION
Human noroviruses are regarded as the single major cause of acute non-bacterial gastroenteritis and foodborne viral illness worldwide but they cannot be cultivated in vitro, and there are no common laboratory animals available for efficient propagation of the viruses. Over the past decades, noroviruses have avoided public attention for being difficult to detect. Recent molecular methods have made the detection, identification and characterization of noroviruses more sensitive and more accurate than before, and have brought new insights into the etiologies of acute gastroenteritis, but the rapid revolution and widespread reported emergence of novel strains of the viruses create an ongoing challenge to the use of molecular techniques to detect the viruses [106]. In addition, given the role that noroviruses play in foodborne and waterborne diseases, research needs to be targeted toward further development, and simplification of these new methods to ensure their broadest applications not only in clinical but also in food and environmental settings.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
The authors thank Michael Kulka for reading the manuscript. The views and opinions expressed here do not necessarily represent the official view of the FDA.

