All published articles of this journal are available on ScienceDirect.

Evaluation in Mouse Model of Combined Virus-bacterial Vaccine Based on Attenuated Influenza A(H7N3) Virus and the Group B Streptococcus Recombinant Polypeptides
Authors Info & Affiliations
Abstract
Background:
Secondary bacterial influenza complications are a common cause of excesses morbidity and mortality, which determines the need to develop means for specific prophylaxis. Group B streptococcal infection is especially common cause of pneumonia among children and the elderly with underlying conditions. Here we investigate in a mouse model the effects of combined intranasal immunization using live attenuated influenza vaccine and recombinant polypeptides based on group B Streptococcus surface proteins.
Methods:
Groups of outbred mice received two doses of the following preparations: 1) the reassortant A/17/Mallard/Netherlands/00/95 (H7N3) influenza virus; 2) a mixture of P6, ScaAB, ScpB1 and Stv recombinant GBS proteins (20 µg total); 3) the A(H7N3) influenza vaccine pooled with the four bacterial peptide preparation; 4) control animals were treated with PBS.
Results:
Intranasal vaccination using LAIV in combination with GBS polypeptides provided advantageous protection against infections with homologous A/Mallard/Netherlands/12/00 (H7N3) wild type virus or heterologous A/Puerto Rico/8/34 (H1N1) followed by serotype II GBS infection. Also, combined vaccination improved bacterial clearance from the lungs of mice.
Conclusion:
Intranasal immunization with LAIV+GBSV was safe and enabled to induce the antibody response to each of vaccine components. Thus, the combined vaccine increased the protective effect against influenza and its bacterial complications in mice compared to LAIV-only.
INTRODUCTION
Infections caused by influenza viruses (the genus Influenzavirus, the family Orthomyxoviridae) increase sensibility of airways to bacterial invasions which often cause severe secondary complications such as bronchitis and pneumonia, especially among young children and the elderly [1-4]. Bacterial pneumonia was the cause of 29% of deaths in the initial period of the A (H1N1)pdm influenza pandemic [5] thus confirmed the socio-economic importance of the influenza bacterial complications. Influenza vaccination can greatly reduce risk of secondary bacterial complications following influenza infection [6]. Despite the increasing importance of associated viral and bacterial infections, viral-bacterial combined vaccine has not yet been developed. In this paper, we attempted to assess the possibility of preventing the mixed virus-bacterial infections in mice using combined intranasal vaccine, including reassortant influenza virus A/17/Mallard/Netherlands/00/95 (H7N3) as a live influenza vaccine (LAIV), and a mixture of recombinant group B streptococcus polypeptides as antibacterial components.
Invasive infections caused by group B streptococcus (GBS) also known as Streptococcus agalactiae are the most common cause of pneumonia in neonates and in the elderly with underlying chronical conditions [7, 8]. Because of the obvious public health ramifications of GBS infection, development of GBS vaccines is highly demanded [9]. One approach of the GBS vaccine development is the use of recombinant polypeptides corresponding to surface bacterial proteins conserved epitopes [10]. The recombinant peptide selection for the bacterial vaccines is based on initial surface bacterial proteins prevalence, conservativeness, immunogenicity and protectivity [11-19].
Previously, immunogenicity and protective efficacy of the recombinant GBS proteins P6 and ScaAB were shown in mice when administered subcutaneously using adjuvants [10]. When administered intranasally to Balb/c mice along with influenza virus possessing deleted NSl-gene, the recombinant GBS proteins demonstrated increased immunogenicity and enhanced protective properties against GBS infection. Because live influenza vaccine development is particularly important for the prevention of respiratory tract infections, we evaluated the combined intranasal vaccine in mouse model based on LAIV and the GBS peptide vaccine (GBSV).
MATERIALS AND METHODS
Viruses. The reassortant A/17/Mallard/Netherlands/00/95 (H7N3) influenza virus (LAIV) containing the surface glycoproteins hemagglutinin and neuraminidase from A/Mallard/Netherlands/12/00 (H7N3) was generated using classical genetic reassortment in 10-day-old developing chicken embryos (CE) [20]. Influenza viruses A/Mallard/Netherlands/12/00 (H7N3) wild type (wt) and A/Puerto Rico/8/34 (H1N1) were obtained from the Virology department collections of viruses, Institute of Experimental Medicine. All viruses were propagated in CE and stored at - 70°C.
Group B streptococcus. S. agalacticae (serotype II) was obtained from collection of the Institute of Experimental medicine (Saint Petersburg, Russia). S. agalacticae were grown in aerobic conditions at 37°С for 18 hours in Todd-Hewitt Broth (THB). Columbia agar with sheep red blood cells were used as a solid medium (Conda Pronadisa, Madrid, Spain).
GBS polypeptides. GBS recombinant polypeptides P6 (30-kDa), ScaAB (35-kDa), ScpB1 (43-kDa) and (Stv130-kDa) were expressed in E.coli and purified as described earlier [10]. All four expressed proteins (with a C-terminal His tag) were obtained in the soluble fraction and purified by immobilized metal affinity chromatography using a Ni-Sepharose column (GE Healthcare, USA).
Immunization. The 8 to-10-week-old female outbred mice were provided by the laboratory breeding nursery of the Russian Academy of Sciences (Rappolovo, Leningrad Region). Four groups of mice (40-60 animals in groups) were lightly anesthetized with ether and intranasally (i.n.) vaccinated with 50 µL divided equally per nostril using the following preparations: 1) 1x106 50% egg infectious dose (EID50) of the A/17/Mallard/Netherlands/00/95 (H7N3) vaccine virus; 2) GBS protein vaccine (GBSV) containing the mix of P6, ScaAB, ScpB1 and Stv recombinant polypeptides (5 µg each, 20 µg total); 3) combined vaccine including 1x106 EID50 of A(H7N3) virus and GBSV; 4) control animals were inoculated by PBS. The mice were immunized twice at an interval of 21 days. To determine vaccine virus reproduction mice were euthanized 3 and 6 days after a single- or mix-inoculation; lung and nasal homogenates were prepared using a disruptor and clarified supernatants were titrated in CE at 34°C to determine infectious virus.
Three weeks after vaccination and revaccination, sera were collected from ether anesthetized mice via submandibular plexus. Nasal secrets were collected from mice after intraperitoneal administration of 0.1 ml of a 0.5% Pilocarpine solution (Sigma-Aldrich, St. Louis, MO, USA) into the tubes containing 0.001 М of serine protease inhibitor phenylmethylsulfonyl fluoride (PMSF). Sera and nasal samples were stored at -20°C.
All procedures involving animals were performed according to the “Rules of Laboratory Practice” Ministry of Health of the Russian Federation Nº 708 n.
Immunogenicity. For hemagglutination-inhibition assay (HI), sera were treated with receptor-destroying enzyme (RDE, Denka Seiken, Tokyo, Japan) as previously described [21] and the HI antibodies against LAIV and against A/PR8/34 influenza virus were quantitated using chicken red blood cells. The enzyme-linked immunosorbent assay (ELISA) was conducted to determine serum IgG and nasal IgA antibodies in 96-well microplаtes (Sarstedt AG & Co, Nümbrecht, Germany) as previously described [21]. For absorption we used 100 HAU/0.1 ml of the whole purified A/17/mallard/Netherlands/00/95 (H7N3) virus or 0.2 mg/0.1 ml of GBSV individual components. The end-point ELISA titers were expressed as the highest dilution that yielded an optical density at 450 nm (OD450) greater than the mean OD450 plus 3 standard deviations of negative controls at an equivalent dilution of sera.
Challenge study. On day 23 after revaccination mice from all groups were intranasally inoculated with 500 50% mouse infectious doses (MID50) of homologous A/Mallard/Netherlands/12/2000 (H7N3)-wt influenza virus (~1x106.5 EID50) or 100 MID50 of the A/PR8/34 influenza virus (~1x102.5EID50). Twenty four hours post infection (p. i.) mice were infected with 1x107 colony-forming units (CFU) of GBS (type II).
Infectious virus titers were detected in the lung homogenates 48, 72 and 96 hours after virus infection in developing CE. Bacterial clearance from the lungs was estimated 2, 24 and 48 hours after GBS infection.
Statistics. Data was processed using Statistica software, version 6.0 (StatSoft, Inc. Tulsa, Oklahoma, USA). Means and standard errors of the means (SEM) were calculated to represent virus titers. Reciprocal antibody titers were expressed as log2 (HI) or as log10 (ELISA) and presented as means±SEM. To compare two independent groups we used a Mann-Whitney U-test. To compare multiple independent groups we used a Kruskal-Wallis ANOVA test. The p-values <0.05 were considered to be statistically significant.
RESULTS
Combined immunization using LAIV and GBSV did not increase the vaccine virus reproduction in the lungs of mice (Table 1).
| Vaccine groups | Day 3 | Day 6 | ||
|---|---|---|---|---|
| Mean virus titers (log10 EID50/ml) ± SEM |
Number infected/total vaccinated | Mean virus titers (log10 EID50/ml) ± SEM |
Number infected/total vaccinated | |
| Virus isolation from lungs | ||||
| LAIV | 2.3±1.1 | 1/4 | 1.5±0.0 | 0/4 |
| LAIV+GBSV | 2.4±1.3 | 1/4 | 1.5±0.0 | 0/4 |
| Virus isolation from nasal turbinates | ||||
| LAIV | 2.6±1.1 | 3/4 | 1.8±0.3 | 1/4 |
| LAIV+GBSV | 2.7±1.2 | 3/4 | 1.5±0.0 | 0/4 |
* - Lungs and nasal tissues were collected 3 or 6 days post infection (p.i.) and titrated in eggs for assessing viral replication. The virus titers are expressed as the mean log10 EID50/ml±SEM from four mice per group. The limit of virus detection was 101.5 EID50/ml. Tissues in which no virus was detected were given a value of 101.5 EID50/ml for calculation of the mean titer. Mice were considered infected if infectious virus was detected in 0.1 ml of 1:10 dilution of tissue homogenate.
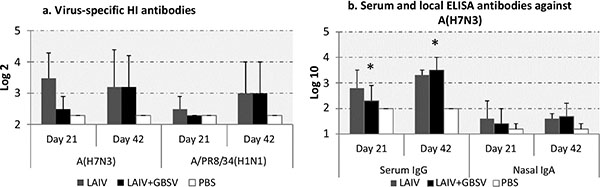
Either single or double immunization with LAIV of A (H7N3) subtype raised serum HI antibodies not only against homologous virus but also against antigenically distinct A/PR8/34 influenza virus (Fig. 1a). Immunization using combined LAIV+GBSV stimulated higher HI antibody titers against A (H7N3) vaccine after revaccination compared to the first vaccination, however the differences were not statistically significant. Serum IgG antibody levels against homologous A (H7N3) influenza virus were significantly increased after revaccination with LAIV+GBSV (p=0.02) whereas double vaccination with LAIV alone did not significantly mount levels of such antibodies. Nasal IgA antibody levels directed to A (H7N3) influenza virus rose only minimally and were similar after combined LAIV+GBSV vaccination and single LAIV immunization (Fig. 1b).
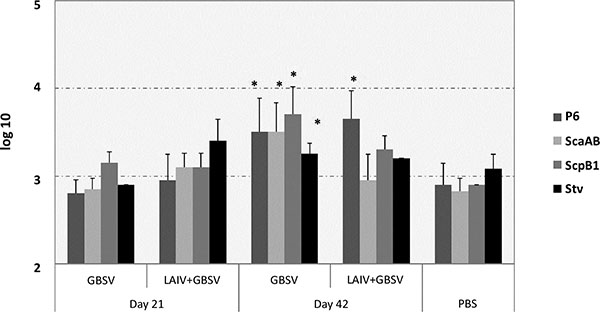
We estimated serum IgG response against each of GBS recombinant polypeptides included in GBSV – the P6, ScaAB, ScpB1 and Stv. Serum IgG levels against P6 polypeptide were increased after the second vaccine dose compared to one dose among either GBSV or LAIV+GBS-vaccinated mice (p=0.003 and p=0.007 respectively). The boost effect against other GBS polypeptides was achieved only in the case of GBSV-based vaccination (Fig. 2). After revaccination with combined LAIV+GBSV vaccine serum IgG levels against ScaAB and Stv were even lower than after primary immunization, although the differences were not statistically significant (Fig. 2). The relatively high levels of serum IgG reacting with GBS polypeptides (> 1:100) among mock immunized animals may be explained by non-specific reaction of serum proteins and changed conformation of the antigen when linked on solid phase [21].
The infectious influenza viruses A (H7N3)-wt or A/PR8/34 reproduced in mouse lungs till at least 96 hours after primary virus challenge which was abused by serotype II GBS infection 24 hours later (Fig. 3). Mice that were immunized with LAIV+GBSV demonstrated significant reduction of A (H7N3)-wt virus titers in the lungs on 96 hours p.i compared to those in the placebo group (p=0.029). LAIV-only vaccinated mice also demonstrated 30 times reduced A (H7N3)-wt virus outcome in contrast to control animals.

Reproduction of distinct A/PR8/34 influenza virus in mouse lungs on 96 hours after infection was reduced due to combined virus-bacterial vaccination (p=0.028) or LAIV immunization, where 4 out of 5 infected animals were completely free from lung virus infection despite additional bacterial load. Perhaps due to an insufficient infectious dose of the A/PR8/34 influenza virus, half of unvaccinated animals in control group (3 out of 6) recovered by 96 hours post virus infection much like 3 out of 5 mice vaccinated with GBS polypeptides.
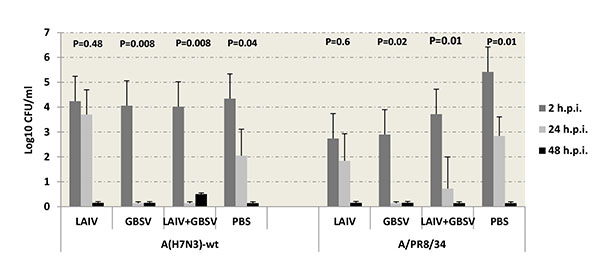
GBS clearance data demonstrated that after A (H7N3)-wt virus infection, the mice immunized with GBSV-only or with LAIV+ GBSV were completely recovered by 24 hours after GBS infection, whereas vaccination with LAIV-only did not reduce GBS titers in the lungs (Fig. 4). In the case of A/PR8/34 primary infection, the mice immunized with LAIV-only demonstrated 500 times reduced bacterial carriage on 2 hour p.i, although on 24 hours p.i. titers of bacteria were similar to those in PBS group. The mice given GBSV vaccine were completely recovered from GBS by 24 hours p.i. (Fig. 4); in LAIV+GBS group 4 mice out of 5 were recovered. On 48 hours p.i. the infectious GBS were not isolated from mouse lungs neither in the case of A (H7N3)-wt nor A/PR8/34 primary infection.
DISCUSSION
This screening study has resolved a number of questions brought up by the need to prepare a nasal associated virus-bacterial vaccine comprising not only live vaccine influenza virus, but also recombinant bacterial polypeptides. In previous studies in mice and ferrets the LAIV based on the reassortant A/17/Mallard/Netherlands/00/95 (H7N3) influenza virus was immunogenic and provided protection against homologous challenge with wild type A(H7N3) virus [22]. Further clinical studies demonstrated that the A (H7N3) LAIV was areactogenic and immunogenic in healthy adult volunteers [23]. Previously, co-administration to Balb/c mice of recombinant GBS peptides and an influenza delta NS1 vaccine, which was unable to reproduce in interferon competent cells and organisms, increased bacterial protein immunogenicity and protective efficacy. In contrast to the previous study, in our work we used a live attenuated virus as an influenza vaccine component. Here we demonstrated that the intranasal administration of LAIV in combination with bacterial GBS peptides was harmless for mice since there was no increase in the vaccine virus reproduction in the lungs. The outbred mice used in our study proved to be an appropriate murine model to assess the vaccination potential of both LAIV and recombinant GBS polypeptides. Firstly, all viruses used in the study such as A/17/Mallard/Netherlands/00/95 (H7N3), A/Mallard/ Netherlands/12/00 (H7N3) and A/Puerto Rico/8/34 (H1N1) reproduced well in the respiratory tract of animals; besides both LAIV and recombinant GBSV were able to induce the immune response after intranasal administration. Previously, McHugh, et al. (2013) used the outbred mice to simulate mild and severe infection caused by influenza A (H1N1)pdm virus together with S. aureus. These two pathogens were considered as the most appropriate model for new vaccines development intended for the human population [24]. Our data suggest that these mice can also be used for the study of viral and bacterial infections caused by other subtypes of influenza virus complicated by GBS infection.
Combined virus-bacterial vaccination improves GBS clearance of the mice’s lungs at 24 hours after bacterial infection following previous A(H7N3)-wt infection as compared to other vaccine groups or non-vaccinated animals while vaccination with LAIV-only decreased bacterial clearance at 24 hours after bacterial super-infection. We can assume that this effect may be related to antibody-dependent cellular cytotoxicity leading to a transient increase in bacterial adherence with complete elimination of streptococci from the lungs by 48 hours. It is a dynamic process and, possibly, after some time in the mock-vaccinated animals with no immune control of virus replication, the destruction of the epithelium will provoke a severe complication of bacterial infection. This data confirm the previously published suggestion of a need for future investigations to better understand the bacterial respiratory pathogen dynamics after LAIV immunization [25].
Previously the development of a GBS vaccine was focused on using the capsular polysaccharide, which is a major virulence factor of GBS. Although due to their chemical structure based on repeating of carbohydrate subunits, microbial polysaccharides often induce only low levels of IgM stimulating little immunologic memory despite repeated immunization [26]. Currently, considerable attention is paid to the study of GBS surface proteins that plays an important role at various stages of infection and participates in the adhesion process. Particularly other virulence factors, including GBS enzymes such as nucleases, proteases, hemolysin or C5a peptidase possessing high immunogenicity due to the direct interaction with the host immune system are currently considered as potential components of vaccines against GBS. In our study the most prominent immune response of serum IgG was obtained against P6 - a recombinant derivative of Bac surface GBS protein. Nevertheless, the fact that intranasal immunization using GBSV-only can result in reduction of bacterial and viral titers in the lungs of mice suggests that antibody response against other GBSV components, which was achieved only in the case of GBSV-based double vaccination, also may play a role in protecting against a viral reinfection followed by bacterial contamination.
CONCLUSION
Thus, our evidence in animal model shows that intranasal immunization with LAIV+GBSV was safe and enabled to induce serum IgG to both viral and bacterial vaccine components. Combined viral and bacterial intranasal immunization using LAIV and recombinant bacterial polypeptides increased the protective effect against influenza and its bacterial complications by reducing primary viral infection and bacterial proliferation that follows.
LIST OF ABBREVIATIONS
| ELISA | = Enzyme-linked immunosorbent assay |
| GBS | = Group B Streptococci |
| GBSV | = Group B Streptococci protein vaccine |
| HAU | = Hemagglutination-inhibition units |
| HI | = Hemagglutination-inhibition assay |
| LAIV | = Live influenza vaccine |
| SEM | = Standard errors of the means |
| Wt | = Wild type |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by the budget of Federal State Budgetary Scientific Institution “Institute of Experimental Medicine”, Saint Petersburg, Russia.
The work was partially supported by grant of the Ministry of Education and Science of Nº 14.613.21.0023.

