All published articles of this journal are available on ScienceDirect.

Comparison of the Bacterial Symbiont Composition of the Formosan Subterranean Termite from its Native and Introduced Range
Abstract
We investigated the bacterial composition in the gut of Formosan subterranean termites (FST), Coptotermes formosanus Shiraki, collected from southern China (native range) vs. Louisiana, U. S. (introduced range) using 16S rRNA gene sequencing. Overall, we identified 213 bacteria ribotypes from thirteen phyla. The enemy release hypothesis could not be invoked to explain invasion success of FST since no pathogens were found among the bacterial gut community regardless of geographic origin. Invasion of new habitats did not significantly change the bacteria composition. Apparently, the tight co-evolutionary link between termites and their gut flora maintains a certain association of species and functional groups. Ribotype richness, bacteria diversity, and proportions of detected phyla were not influenced by geographic origin of FST samples; however, these parameters were affected by storage of the samples. Ethanol storage of termite samples (5 yrs) increased the relative proportions of gram-positive bacteria versus gram-negative bacteria.
INTRODUCTION
Intestinal symbionts have contributed to the evolutionary success of various insects [1, 2]. Gut symbionts enable a variety of insects to utilize refractory and nutrient-deficient diets by supplying digestive and detoxifying enzymes and serving as sources of nitrogen, vitamins and energy. Symbionts can also protect their host from invasion by foreign pathogens [1].
Because there are so many benefits conferred by symbiosis, questions have been raised about whether symbionts could facilitate the establishment and success of invasive insects by enabling their hosts to tolerate rapid changes in habitat and diet. For example, it has been shown that the intestinal symbiont flora of insects adapts quickly to changes in environmental conditions and diet by the induction of enzymes or a shift in the species composition [1, 3-7]. Also, symbionts can be digested by the host and used as a temporary protein, lipid, carbohydrate and vitamin source during times of starvation [8]. While the partnership with beneficial symbionts that are obligate for survival has to remain intact through the invasion process, transient pathogens might get lost and thus increase the survival and competitive edge of the introduced populations (enemy-release-hypothesis [9]). Therefore, investigating both, the composition of beneficial symbionts and pathogens in the native and introduced range of an invasive species might reveal cues to its invasion success.
Symbiont communities have been investigated in only a few invasive insect species, including the Asian longhorned beetle, Anoplophora glabripennis [10], the gypsy moth, Lymantria dispar [11], the emerald ash borer, Agrilus planipennis [12] and the invasive subterranean termite species Coptotermes formosanus [13, 14]. Although such invasive species/symbiont complexes have been described for certain locations or populations, without direct comparison of the symbiont communities in the native and introduced ranges, it remains unknown how the symbiont community changes and adapts as the hosts invade new regions and if there is a release from pathogenic members in the symbiont community in the introduced range.
Invasive subterranean termites of the genera Reticulitermes and Coptotermes (Isoptera: Rhinotermitidae) and their diverse symbiont community have a large impact on ecology and economy in tropical, subtropical and temperate regions around the world due to their ability to degrade plant material and cycle nutrients. Plant material is rich in lignocelluloses, but deficient in vitamins and essential components for protein and fat synthesis. Therefore, subterranean termites are dependent on a diverse assemblage of eukaryotic (Protista, Fungi) and prokaryotic (Archaea, Eubacteria) symbionts in their hindguts to digest lignocellulosic compounds [6] and supplement their nutrition with nitrogen [15, 16], acetate for energy production [17] and vitamins [18, 19].
While the number of eukaryotic symbiont species in the subterranean termite gut is relatively limited, the prokaryote diversity is astonishing and only a small fraction has been cultured so far [20]. To obtain comparatively unbiased bacterial inventories, culture independent molecular methods currently are used to identify species, describe species richness, relative abundance, phylogenetic affiliation and putative functions (reviewed in [21]). The most widely used approach to describe bacterial communities is to sequence species-specific 16S rRNA genes. Certain regions in rRNA genes accumulate variation as the organisms diverge. Flanking regions remain conserved and can therefore be used to construct universal primers for amplification and sequencing of the rRNA gene [22]. Bacteria inventories in subterranean termite guts have been described using 16S rRNA sequences and culture-independent methods for Reticulitermes speratus from Japan [23-26], R. flavipes from North America [27], R. santonensis, the European synonym of R. flavipes [28], and the Formosan subterranean termite (FST), C. formosanus from Japan [14]. The latter species is one of the most successful invasive insect pests known and continues to spread in tropical and subtropical regions of Asia, the Pacific region and southern United States. The FST has been transported from its native range, most likely southern China [29], to Japan prior to the 1600s, to Hawaii in the late 1800s and to the continental United States in the late 1950s [30]. In Louisiana, FST was first documented in 1966, but was probably introduced earlier [31]. As a globally successful invasive species with a diverse symbiotic bacterial assemblage, the FST is a good model for the study of the effects of introduction upon the gut microbiota. To date, only the symbiont community of FST in its introduced range has been studied [13, 14], and the range of variation of the symbiont communities among FST colonies has not yet been assessed from either the same or from different geographical regions.
Comparisons of the symbiont community among FST colonies from native and introduced populations is expected to reveal (1) which symbionts are obligate (i.e., they are present regardless of the geographic origin of the termite colonies), (2) if introduction changes the bacterial composition and/or (3) if there is a reduced pathogen load in introduced populations. Therefore, we used 16S rRNA gene sequencing to describe and compare the composition of bacterial communities in multiple FST colonies from native populations in China and introduced populations in Louisiana, U.S.A. (described in this study) as well as in Japan (data reanalyzed from [14]). Part of the FST samples from Louisiana and China had been preserved in ethanol for about 5 yrs. While 95% ethanol has been shown to be capable of preserving the DNA of insects, often for years [32], the DNA preservation of the insect endosymbionts has not yet been well studied. Short-term storage of termite material in 95% ethanol at room temperature did not lead to changes in the bacteria biodiversity [13, 33]; however, no data were available for storage longer than a couple of weeks or months. Therefore, we also investigated if long-term ethanol storage may change the detectable bacteria composition.
MATERIALS AND METHODOLOGY
Termite Collection
Three FST colonies from Louisiana, U.S.A., were collected in the summers of 2005, 2004 and 2003, respectively: sample 1 from the city of Lake Charles, sample 2 from Chalmette Battlefield, New Orleans and sample 3 from City Park, New Orleans. The guts of 50 FST workers per colony were dissected on the day of collection (see below). These samples (1-3) are referred to as LA fresh. Additional workers from the FST colony from City Park were subsequently preserved in 95% ethanol (Sigma-Aldrich Corp., St. Louis, MO, U.S.A.) for 5 years at room temperature until DNA extraction was performed in 2008 (sample 4). Two additional FST colonies were obtained that were collected in fall 2001 from Louis Armstrong Park (sample 5) and the French Quarter (sample 6) in New Orleans and stored in 95% ethanol at room temperature until dissection in 2006 (5 years). Samples 4-6 are referred to as LA EtOH. Three FST colony samples from China, collected in 2001 from Hunan Province (sample 7), Sanshui (sample 8) and Zhongshan (sample 9) (both from Guangdong Province) were dissected after 4-5 years of storage in 95% ethanol in 2005 and 2006. These samples (7-9) are referred to as China EtOH.
DNA Extraction and Sequencing
For total DNA extraction, 50 worker guts were extirpated from each sample and homogenized in 200 μl of TE buffer (10mM Tris-HCl, 1mM EDTA, pH 8.0) under sterile conditions [7]. After centrifuging the mixture at 5000 × g for 10 minutes, the pellet containing the bacteria was resuspended in 200 μl lysis buffer (20mM Tris-HCl (pH 8.0), 2mM sodium EDTA, 1.2% Triton X-100, 20 mg/ml lysozyme) and incubated overnight at 37oC. The remaining DNA extraction procedure was performed using the Qiagen DNeasy Tissue Kit (Qiagen, Valencia, CA). DNA concentration was quantified using the Thermo Scientific NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, U.S.), and the quality of the DNA was determined using gel electrophoresis.
The 16S rRNA genes of the gut bacteria were amplified using the bacteria-specific primer pair 27F (5’-AGAGTTT-GATCCTGGCTCAG-3’) and 1492R (5’-GGTTACCTTG-TACGACTT-3’ [34]) in reactions consisting of 1X PCR buffer (60mM Tris-HCl, 15 mM (NH4)2SO4, pH 8.5 and 2.5mM MgCl2), 0.25 mM dNTP, 1U AmpliTaq DNA polymerase (Roche, Indianapolis, IN, U.S.), 0.25 μg of each primer with approximately 10 ng of DNA template from the fresh FST samples and 30-160 ng from the ethanol-stored FST samples. The dNTPs were added after heating the PCR reaction tubes for 2-3 min at 80oC. Reaction mixtures were incubated in a PTC-200 thermal cycler (MJ Research, Reno, NV, U.S.) using the following program: 94oC for 2 min; 25 cycles of denaturation (94oC for 1 min), annealing (55oC for 2 min) and extension (72oC for 3 min); and a final 7 min extension at 72oC. PCR products were purified using the UltraClean PCR Clean-Up kit (MoBio, Solana Beach, CA, U.S.) and cloned using the TOPO TA Cloning kit (Invitrogen, San Diego, CA, U.S.). The clones were grown overnight on selective LB media and clones with DNA inserts were selected through blue-white screening. Clones with DNA inserts were bi-directionally sequenced at the Interdisciplinary Center for Biotechnology Research (ICBR) at the University of Florida (Gainesville, FL, U.S.). The full-length 16S rRNA gene sequences were compared to the GenBank/EMBL/DDBJ databases using BLAST and classified into different ribotypes using the < 97 % sequence similarity criterion [35]. Chimeric sequences detected using Bellerophon [36], ChimeraCheck [37], Mallard [38], signature analysis and BLAST results were excluded from further analyses. Ribotypes were submitted to the GenBank/EMBL/DDBJ databases under the accession numbers GQ502463-2668.
Classification of Ribotypes and Phylogenetic Analysis
Using the default settings on the naïve Bayesian rRNA classifier [39] on the RDP website [40], the sequences were classified into the known major bacteria lineages based on a confidence level of 80%. The ribotypes were named according to the prefix Cf (Coptotermes formosanus) followed by a first numeral indicating the phylum classification of the ribotype on the RDP website and a second numeral assigned to each ribotype. Classification of the sequences was confirmed using BLAST and reclassified based on the latest publications. Sequence divergence among the ribotypes was calculated using the Kimura 2-parameter model. Phylogenetic trees were constructed using the neighbor-joining method as implemented in MEGA 4.0 with 1000 bootstrap replicates (Fig. 1).
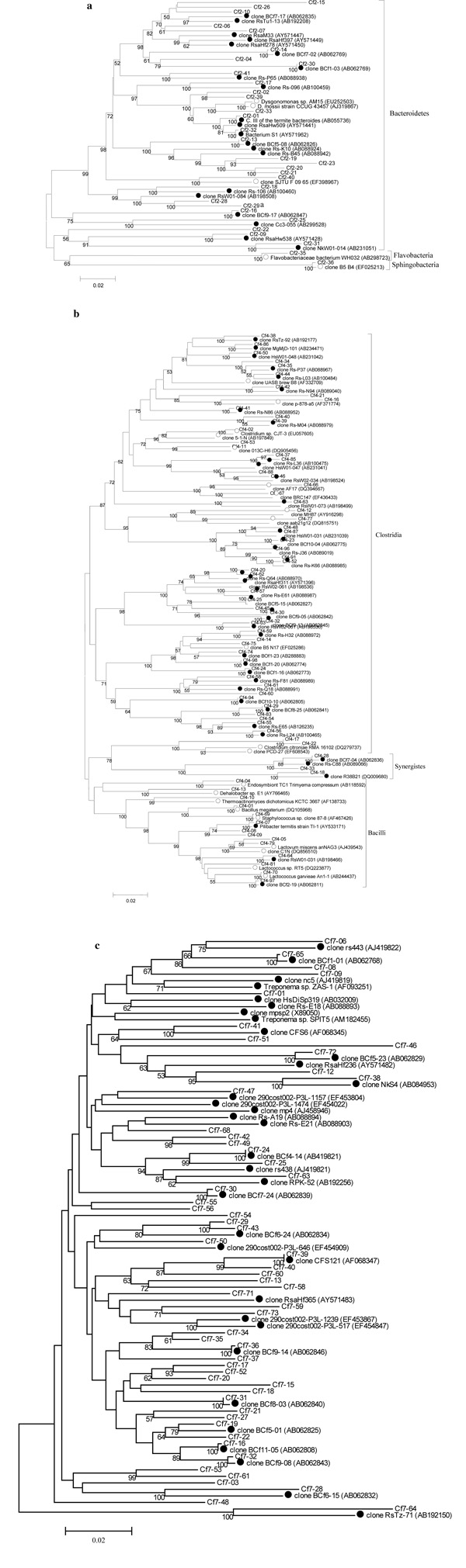
Phylogenetic trees constructed from the 16S rRNA gene sequences of gut bacteria of Formosan subterranean termite (FST) samples from Louisiana and China (Cf) and their highest match from the DDBJ/EMBL/GenBank databases. A. Bacteroidetes, B. Firmicutes, C. Spirochaetes, D. Actinobacteria, E. Proteobacteria, F. TM7, G. Verrucomicrobia and Planctomycetes. The phylogenetic trees were constructed using the neighbor joining method with 1000 bootstrap replicates. Only bootstrap values of ≥50 are indicated on the branch nodes. The scale bars represent 2% difference in nucleotide sequence. Closely related ribotype sequences from other studies were included for comparison and marked with circles (● termite-specific bacteria, ○ environmental bacteria). A. Bacteroidetes. B. Firmicutes & Synergistes. C. Spirochaetes. D. Proteobacteria. E. Actinobacteria. F. TM7. G. Planctomycetes & Verrucomicrobia.

Rarefaction curves of bacterial phyla and ribotypes depending upon the number of FST samples. Observed richness of phyla and ribotypes (Sob) was measured based on the Mao Tau index. Total richness was estimated using Chao 1. Upper and lower 95% confidence intervals are given.
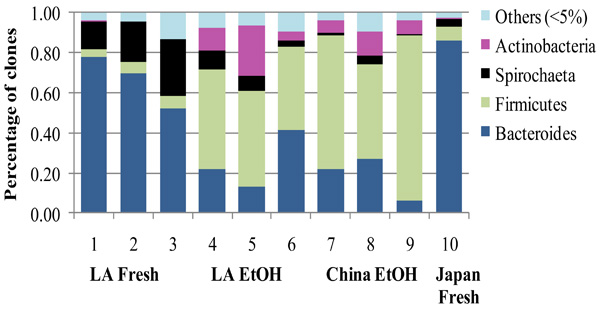
Proportions of the four major phyla (>5%) and other minor phyla (<5%) in fresh and EtOH-stored FST samples from the native range in China and the introduced range in Louisiana (LA). Previously published data from an FST sample from Japan were included for comparison [14].
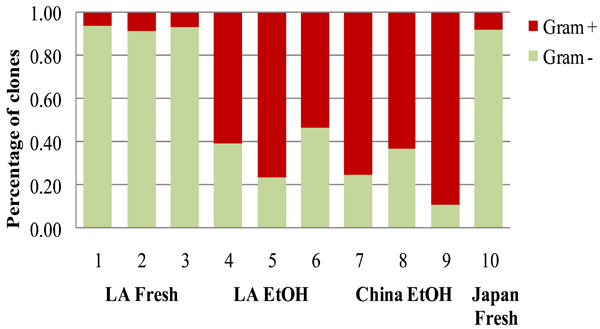
Proportions of gram positive and gram negative bacteria in fresh and EtOH-stored FST samples from the native range in China and the introduced range in Louisiana (LA) and Japan [14].
Bacterial Ribotype Richness, Diversity and Similarity Analysis
Rarefaction analyses were performed for bacterial communities from each of the FST colonies, averaged over 100 randomization runs using the EstimateS V7.5 software [41]. Mao Tau [42] was used to measure the observed ribotype richness in each FST colony. To estimate the percentage of total ribotypes captured in each colony, the non-parametric estimators of expected ribotype richness based on the numbers of singletons and doubletons in each sample (Table 1), Chao1 [43], ICE [44], ACE [45] and Jack1 [46] were used. The Simpson’s index of diversity (1-D), which is influenced by abundance since it reflects the likelihood that two ribotypes randomly drawn from the same sample are different, and Shannon index, which weighs both the ribotype richness and abundance and has a scale between 0 (low diversity) and ~4.6 (high diversity), were used as a measure of the ribotype diversity. The similarity of bacterial composition among the FST colonies was determined using the Chao-Jaccard Abundance [47], Bray-Curtis [48] and the Morisita-Horn indices [49]. All indices showed similar results. Therefore, only data based on Morisita-Horn are presented. The richness, diversity and similarity indices for each of the FST colonies were interpolated using rarefaction analysis to be able to compare all samples to the sample with the lowest number of clone samples (123 clones, sample 4, Table 1) to avoid biases due to unequal sample sizes. Effects of geographical origin of the FST samples (introduced vs. native range) and storage condition (fresh vs EtOH-stored) on ribotype richness, diversity, similarity of bacteria composition among samples and bacterial phylum proportions were tested using General Linear Model (GLM) analyses; differences were considered significant if two-tailed Mann-Whitney U test and Kruskal-Wallis ANOVA showed p < 0.05 (SPSS 17.0, SPSS inc., Chicago, IL).
General Information about the Number of Clones and Ribotyes, Estimated Percentage of Ribotypes Captured and Bacteria Diversity in Formosan Subterranean Termite (FST) Samples from Louisiana and China. For Comparison, the Same Indices were Calculated Based on Previously Published Data for one FST Colony from Japan [14]
| LA Fresh | LA EtOH | China EtOH | Japan | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FST samples | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| General information | ||||||||||
| Clones sequenced | 350 | 248 | 276 | 123 | 161 | 177 | 172 | 238 | 131 | 250 |
| Ribotype richness | 57 | 51 | 75 | 40 | 48 | 42 | 38 | 54 | 23 | 49 |
| Singletons (%) | 41(11.7) | 35(14.1) | 49(17.8) | 33(26.8) | 30(18.6) | 17(9.6) | 19(11.0) | 34(14.3) | 12(9.2) | 34(13.6) |
| Doubletons (%) | 7(2.0) | 8(3.2) | 9(3.3) | 3(2.4) | 8(5.0) | 8(4.5) | 9(5.2) | 7(3.0) | 5(3.8) | 7(2.8) |
| Uniques (%) | 19(33.3) | 9(17.6) | 18(24.3) | 12(30.0) | 12(25.5) | 17(9.6) | 4(9.5) | 11(28.9) | 15(27.8) | 25(51.0) |
| Total ribotype richness (% ribotypes captured) | ||||||||||
| Chao1 | 33-100 | 32-95 | 48-100 | 41-79 | 45-93 | 33-67 | 30-66 | 38-90 | 18-42 | 30-92 |
| ACE | 8-100 | 18-91 | 40-100 | 38-71 | 39-84 | 27-55 | 26-55 | 24-87 | 12-36 | 13-86 |
| ICE | 8-100 | 18-91 | 40-100 | 38-71 | 39-84 | 27-55 | 26-55 | 24-87 | 12-36 | 36 13-86 |
| Jack 1 | 33-48 | 35-49 | 51-68 | 45-59 | 49-64 | 35--48 | 32-45 | 41-55 | 19-28 | 32-46 |
| Bacteria diversity | ||||||||||
| 1-Simpson | 0.49 | 0.56 | 0.81 | 0.94 | 0.95 | 0.88 | 0.89 | 0.89 | 0.76 | 0.51 |
| Shannon | 1.46 | 1.66 | 2.60 | 3.11 | 3.25 | 2.71 | 2.64 | 2.72 | 1.91 | 1.50 |
Estimators of ribotype richness (Chao1, ICE, ACE and Jack1) and their 95% confidence intervals were used to calculate the percentage range of total ribotypes captured in each sample.
The Simpson index of diversity and Shannon index were used as a measure of the ribotype diversity. Rarefaction curves were used to interpolate total estimated richness and bacteria diversity for each of the FST samples to compare to the sample with the lowest number of sequenced clones (123 clones, sample 4). Note that sample 3 and 4 are from the same FST colony, but different storage conditions.
Similarity of the Bacteria Composition among FST Samples from the Same or Different Storage Condition (fresh vs. EtOH) and Geographical Region (Native Range: Southern China, Introduced Range: Louisiana (LA) and Japan [14]
| FST Colony | LA Fresh | LA EtOH | China EtOH | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| LA Fresh | 1 | - | ||||||||
| 2 | 0.995 | - | ||||||||
| 3 | 0.867 | 0.893 | - | |||||||
| LA EtOH | 4 | 0.329 | 0.344 | 0.432 | - | |||||
| 5 | 0.237 | 0.248 | 0.342 | 0.590 | - | |||||
| 6 | 0.741 | 0.764 | 0.885 | 0.592 | 0.542 | - | ||||
| China EtOH | 7 | 0.366 | 0.376 | 0.468 | 0.440 | 0.728 | ||||
| 8 | 0.555 | 0.573 | 0.695 | 0.565 | 0.743 | 0.806 | 0.795 | - | ||
| 9 | 0.089 | 0.090 | 0.100 | 0.321 | 0.467 | 0.170 | 0.349 | 0.546 | - | |
| Japan | 10 | 0.995 | 0.994 | 0.878 | 0.339 | 0.237 | 0.755 | 0.372 | 0.565 | 0.089 |
The Morisita-Horn similarity index was used to determine the fraction of shared ribotypes
Lighter shading – same storage condition or geographic region.
Darker shading – different storage conditions or geographic regions.
RESULTS
Bacteria Species Inventory
A total of 1,876 clones containing 16S rRNA genes from gut bacteria of nine FST samples from China (native range) and Louisiana (introduced range) were sequenced. Twenty-four ribotypes consisting of 52 clones were determined to be artifact sequences and were excluded from subsequent analyses. The remaining 213 bacteria ribotypes comprising 1,824 clones were classified into 13 bacteria phyla: Bacteroidetes (42.9% of the total clones), Firmicutes (30.5%), Spirochaetes (11.3%), Actinobacteria (5.7%), Proteobacteria (2.2%), Tenericutes (1.6%), candidate division Termite Group 1 (1.0%), candidate division TM7 (0.6%), Verrucomicrobia (0.6%), Planctomycetes (0.5%), candidate division Synergistes (0.2%), candidate division ZB3 (0.1%) and Cyanobacteria (0.1%) (Fig. 1A-G).
The most abundant ribotype, Cf2-30 (706 clones, 38.71% of the total clones analyzed), was classified within the phylum Bacteroidetes (Fig. 1A). The most diverse phylum was the Firmicutes, with 72 bacteria ribotypes belonging to Clostridia and Bacilli (Fig. 1B). The second and third most abundant ribotypes, both belonged to the class Clostridia (Cf4-32: 147 clones, 8.10% of total clones; Cf4-07, 125 clones, 6.85% of the total clones). The remaining ribotypes occurred in much smaller proportions, each comprising less than 1.92% of the total clones.
Of the 213 total ribotypes, 151 (71%) were considered novel with less than 97% sequence similarity to 16S rRNA gene sequences in the DDBJ/EMBL/GenBank databases. These novel ribotypes accounted for 30% of the total clones. The remaining 62 ribotypes (29%) had sequence similarities of 97% or higher to genes in public databases. Of those, 33 ribotypes (64% of total clones) were previously identified from the guts of C. formosanus (including the three most abundant ribotypes, Cf2-30, Cf4-32 and Cf4-07, mentioned above), 11 ribotypes (3% of total clones) were from other termite guts and the remaining 18 ribotypes (3% of total clones) were endo- or ectosymbionts of other organisms or from the environment. The bacterial ribotypes were not clustered according to the storage conditions of the FST samples (fresh vs. alcohol) or the geographic region of the FST samples (Louisiana vs. China) in the phylogenetic trees.
The ribotype richness and bacterial diversity of each FST sample was interpolated for the lowest clone sample size (123, sample 4) using rarefaction analysis to avoid biases due to unequal sample sizes (Table 1). Ribotype richness per FST sample ranged from 23-74. However, the estimated total richness is likely to exceed the observed ribotype richness (Table 1). Bacterial diversity estimated via the Simpson index of diversity ranged from 0.49 to 0.95 per sample. The Shannon index ranged from 1.46 to 3.25 (Table 1). A considerable proportion of ribotypes in each FST sample ( ≥ 9.5 % Table 1) were unique, i.e. only found in one FST sample.
The rarefaction curve of observed number of phyla depending on the number of FST samples levels off and approaches the curve of estimated total number of phyla (Fig. 2). This indicates that 10 FST samples represent sufficient sampling effort and that the 13 phyla observed capture the majority of bacterial phyla expected in the guts of FSTs. The continued slight incline of the accumulation curve of the number of ribotypes, however, suggests that more ribotypes might be discovered if further FST samples would be analysed. The 213 ribotypes represent at least 55% to 77% of the estimated total number of ribotypes (Fig. 2).
Effects of Geographic Origin and Storage of the FST Samples
Ribotype richness, diversity and proportions of bacteria phyla showed a wide range among samples (Table 1), so we investigated whether this variation could be explained by geographical origin (introduced vs. native range) or storage of the FST sample (processed immediately vs. stored in EtOH).
Observed ribotype richness was not affected by geographic origin (native vs. introduced range) of the samples (p = 0.17, F = 2.345, df = 1, 8, GLM). However, ribotype richness was significantly higher in fresh samples from LA and Japan (data reanalyzed from [14]) than in EtOH-stored samples from LA and China (p = 0.038, n = 10, U = 2.000) and marginally higher in LA fresh samples than in LA EtOH samples (p = 0.10, n = 6, U < 0.001, two-tailed Mann-Whitney U-test). The ribotype richness of two samples from the same FST colony, one (sample 3) processed immediately after collection and the other (sample 4) processed after 5 years of alcohol storage showed a reduction of the ribotype richness of almost 50% (from 74 to 40 ribotypes, Table 1).
Similar to richness, bacteria diversity measured by the Simpson and Shannon indices was not affected by geographical origin (p > 0.20, df = 1, 8 for both indices, GLM). However, bacteria diversity was significantly affected by storage condition (Simpson: p = 0.003, F = 18.535; Shannon: p = 0.021, F = 8.259, df = 1, 8, GLM). The bacterial diversity in the fresh samples from LA and Japan was significantly lower than the diversity of the EtOH-stored samples from LA and China (for both 1-D and Shannon: p = 0.019, n = 10, U = 1.00). When comparing the bacterial diversity of samples from LA only, fresh samples still showed marginally less diversity than EtOH-stored samples (p = 0.10, n = 6, U = 0.001, two-tailed Mann-Whitney U-test).
Morisita-Horn similarity values showed that the proportions of shared bacteria of FST samples from within the native range and within the introduced range were not different from the proportion of shared bacteria among the native and introduced range, i.e., the intercolonial variability of the bacterial inventory, was not significantly different within and among regions (p = 0.478, U = 13.00, Mann-Whitney U, Table 2). However, similarity in bacterial composition among samples was dependent upon whether samples came from the same or different storage conditions (fresh vs EtOH-preserved) (p = 0.001, F = 12.654, df = 1, 43, GLM). More bacterial species were shared among the fresh Louisiana FST colonies and among the ethanol-stored Louisiana FST colonies compared to those that were shared between the fresh and ethanol-stored Louisiana FST colonies (p = 0.025, U = 5.000, two-tailed Mann-Whitney U-test) (Table 2). A low proportion of shared species indicates that the bacterial communities found in the fresh FST colonies were somewhat distinct from the ethanol-stored FST colonies.
Proportions of each of the major phyla (Bacteroides, Spirochaetes, and Actinobacteria were not significantly affected by geographical origin of the FST samples (native vs. introduced range, p > 0.20, F < 1.32, df = 1, 7). Only the Firmicutes were marginally affected by geography (p = 0.05, F = 5.577, df = 1, 7, GLM). However, the proportions of the Bacteroides (p = 0.003, F = 19.914), Firmicutes (p = 0.001, F = 30.621) and Actinobacteria (p = 0.022, F = 8.554, df = 1,7, GLM) were significantly affected by storage condition. Storage did not significantly affect the Spirochaetes as long as the previously published low spirochaete proportion of the Japanese sample [14] was included in the analysis. However, in a separate analysis of FST samples from LA and China (excluding Japan), the Spirochaetes were significantly affected by storage condition (p = 0.017, F = 10.714, df = 1, 6).
The proportion of the most abundant phylum Bacteroides, was significantly higher in the fresh samples than in the EtOH-stored samples (p = 0.01, Chi-Square = 6.585, df = 1, Kruskal-Wallis ANOVA, Fig. 3). The Spirochaetes were marginally more abundant in fresh samples (p = 0.05, Chi-Square = 3.70, df = 1). On the contrary, the gram-positive phyla Firmicutes and Actinobacteria were significantly more abundant in the EtOH stored samples than in fresh samples (p = 0.01, Chi-Square = 6.59, df = 1 for both phyla). When LA samples were considered separately from the other geographic locations the proportions of Firmicutes and Actinobacteria were still significantly higher in EtOH-stored samples (p = 0.04, Chi-Square = 3.97, df = 1) while Bacteroides and Spirochaeta were marginally higher in fresh samples (p = 0.05, Chi-Square = 3.85, df = 1, Fig. 3).
When all phyla (including the minor phyla of < 5% representation) were assorted according to their cell wall characteristics, storage condition affected the proportions of gram negative vs. gram positive bacteria in the samples (p < 0.001, F = 61.257); however, there was no effect of geographic origin (p = 0.16, F = 2.459, df = 1, GLM). The proportions of gram-negative bacteria, which were predominant in the fresh samples, were reduced in the EtOH-stored samples, while the proportion of gram positive bacteria increased in the EtOH-stored samples (all samples: p =0.01, Chi-Square = 6.55; LA only: p = 0.05, Chi-Square 3.86, df = 1, Kruskal-Wallis ANOVA, Fig. 4).
DISCUSSION
We compared the bacterial composition in the guts of Formosan subterranean termites from multiple samples across the native (southern China) and introduced range (Louisiana, U. S. and Japan) using culture-independent 16S rRNA sequencing. Since it is often not practicable to ship live termite samples due to strict import/export regulations and also because isolation of termites from their natural environment rapidly changes the gut bacteria composition [7], we also investigated how prolonged storage in ethanol would affect the detection of ribotypes.
Data from our study using multiple colonies across geographic regions showed an increase in the number of ribotypes identified in the FST hindgut of more than four-fold compared to previously published studies [13, 14]. We identified a total of 213 different ribotypes from eight FST colonies across the native (southern China) and the introduced range (Louisiana, U.S.A.), while the previous study by Shinzato et al. [14] found 49 ribotypes and 9 phyla in one colony from Japan. All of the ribotypes from the Japanese FST colony were also found in colonies from Louisiana and China plus an additional 164 ribotypes. The majority of the bacteria were novel, but closely related to bacteria previously found in the guts of subterranean termites, supporting the hypothesis of termite-specific lineages [13, 14, 21, 23-25, 27].
The most abundant ribotypes were shared among FST colonies regardless of their geographic origin, which suggests that these particular ribotypes fulfill important roles within the gut of FST. The predominant ribotype in most of the Louisiana and China FST samples (Cf2-30) was a Bacteroides species identical to BCf1-03 described previously from a Japanese FST colony [14, 50]. This Bacteroides species is an endosymbiont of Pseudotrichonympha grassii, a flagellate protozoan found only in the guts of the FST [50]. Complete genome analysis of this endosymbiotic bacterium has revealed genes for sugar fermentation and carbohydrate storage, but also nifH genes, which predict the ability to fix nitrogen [51]. The second most abundant ribotype, Cf4-32 from the Clostridia class, was highly similar to BCf9-13 from the Japanese study [14]. Many clostridia degrade polysaccharides to produce acetone, alcohol, acetate, lactate, CO2, and hydrogen [52-55] and others can ferment nitrogenous or lipid compounds [60]. Acetogenic clostridial species are one of the major groups in termites based on culture-independent studies [14, 23, 25, 28, 56, 57].
The third most abundant ribotype, Cf4-07, was almost identical (99%) to Pilibacter termitis, a lactic acid bacterium that we previously cultured and described from a FST colony from Hawaii [58] and BCf6-17 in the Japanese study [14]. As a lactic acid bacterium this species is likely to be involved in sugar fermentation and pH regulation of the gut [59]. Aerotolerant lactic acid bacteria capable of efficient O2-reduction also provide microoxic zones for the strictly anaerobic microorganisms in the gut [58, 61, 62]. Several lactic acid bacteria have been shown to recycle carbon and nitrogen by metabolizing uric acid [15].
The enemy-release hypothesis [9], which would have explained invasion success of FST due the loss of pathogen pressure following introduction to a new habitat, was not applicable to the gut bacteria community. Only a small percentage of the 213 ribotypes identified in the FST gut (8 %) belonged to bacteria other than from termite-specific lineages and none resembled known pathogens, regardless whether the FST samples originated from the native or the introduced range of FST. As suggested before, the highly adapted gut microflora apparently protects the termite gut from invasion by pathogens and transients [1, 63]. Therefore, there was no support for our original assumption that the FST populations in China could be used as a resource for identifying potential bacterial biocontrol agents.
Ribotype richness, bacterial diversity, similarity of bacterial communities among FST samples, and proportions of phyla were not affected by the geographic origin of the FST samples, whether FST were collected from their native (China) or introduced range (Louisiana in the U.S.A. and Japan [14]). This was surprising, because we expected that introduction to a new habitat would either (a) reduce richness and diversity of bacteria in a termite colony due to a founder effect or, alternatively, (b) increase richness and diversity by incorporating bacteria from the new environment and thus (c) impact the similarity among colonies and the proportions of phyla.
- Previously, we recorded evidence of founder effects and genetic bottlenecks in FST populations from Louisiana [64, 65], which were attributed to the introduction of a limited number of FST colonies that established populations in Louisiana [66]. The genetic diversity was significantly reduced in termite colonies from the introduced range (U.S.A. mainland, Hawaii, and Japan) compared to those from the native range (China) [67-70]. Therefore, we expected to detect a similar bottleneck effect reflected in reduced symbiont diversity in the introduced range of FST. However, richness, diversity, and bacterial phylum composition did not differ between FST samples from the native and introduced range. Apparently, in such a complicated network of multilevel symbiosis as that of the termite gut, it is necessary to retain a certain number and composition of different bacterial species and phyla to support the balance and fulfill all the important functions of termite nutrition and gut ecology. Although only a limited number of termite colonies have been introduced to the United States [65], each colony had to have travelled with a sufficient “package” of bacterial species in the guts of their workers in order to survive. The package consists mostly of termite-specific lineages (>70% of the known ribotypes were only found in termites), which are unable to survive outside the termite gut, but were obligatory for termite survival [14, 25, 71]. Therefore, bacteria diversity was not reduced in the introduced range.
- If transient bacteria had been picked up after introduction to a new environment, we would expect to find an increased diversity in the introduced range. However, there is only a minute fraction of environmental bacteria present in the termite gut (8% of the ribotypes and 3% of total clones in our study). While some other social insect species may harbor mainly environmental bacteria in their guts (e.g. red imported fire ants [72] and pharaoh ants [73]), termite gut microorganisms are coadapted and highly specialized symbionts and the bacterial community protects the termite gut against invasion by foreign bacteria [1, 63]. Therefore, transient bacteria have no detectable impact on the overall bacteria richness and diversity or major phyla composition within FST colonies.
- Given the recent introduction of a limited number of FST colonies to similar urban habitats in Louisiana [65], we also would have expected that the bacterial communities from colonies from Louisiana would share more ribotypes among each other than are shared between colonies from Louisiana and colonies from China. However, there was no detectable difference in the intercolonial variability of bacterial communities from FST colonies from the same geographical range when compared to those from different geographical ranges. Although ribotype richness and diversity within FST colonies were similar, the proportion of shared bacterial species varied considerably among FST samples, even when colonies from the same geographical region were compared (Table 2). Field studies on colonies of Hodotermes mossambicus and several Reticulitermes and Microcero termes species, confirmed that the bacterial community of termite colonies usually differs considerably between sampling sites, even if colonies were sampled from the same geographical region [71, 74].
Since the ribotype richness and diversity is high within FST colonies, even a few introduced FST colonies could have carried hundreds of bacteria species. Each termite colony regardless of geographic origin has to contain all the obligatory functional groups of symbionts to support the energy and nutritional needs of the colony. These symbionts, however, do not necessarily have to be identical species, but could consist of closely related species or species with overlapping functions. For example, sequencing of nitrogen fixation genes (nifH) from the mixed bacterial population in the gut of termites provided evidence for the presence of an unexpected diversity of nitrogen fixing bacteria and nitrogenases in the guts of Reticulitermes and Coptotermes species [16, 24]). Acetogenesis is performed by protozoa, various spirochaetes and clostridia-related bacteria, among others [24, 75, 76].
Given the considerable bacterial diversity in termite populations, intercolonial variation could be explained in two ways. First, the bacterial community of each newly founded colony is expected to be a unique mixture of bacteria provided by the founding pair of reproductives, which likely carry different bacterial compositions since they in most cases originated from different colonies [77, 78]. Second, once colonies are established, adaptation to different habitat conditions and diet would further shift the composition of the bacterial community. This shift is due to changes in proportions of bacteria established in the gut and does not involve significant uptake of bacteria from the environment (see above). It has been shown that the relative proportions of bacterial groups in termite laboratory colonies rapidly shift in response to changes in rearing conditions or diet [7]. Bacterial communities in the FST were shown to differ by 60% when comparing FST colonies fed with high molecular weight carbon sources and those fed with low molecular weight carbon sources [6]. Within the gut of the wood-feeding higher termite Nasutitermes takasagoensis, spirochaetes were predominant in the wood- and wood powder-fed termites, Bacteroidetes were predominant in thexylan-, cellobiose- and glucose-fed termites, while Firmicutes were predominant in the xylose-fed termites [79].
The variances among the bacterial communities of FST colonies were not likely due to transient bacteria, which represent only a minute fraction of the ribotypes (see above), nor caused by partial sampling. The variability of bacterial composition among members of the same termite colony is likely low [74], because they exchange bacteria via trophallaxis and the obligatory refaunation after molting [80, 81]. Rarefaction analyses showed that the number of termite colonies sampled was sufficient to detect most of the phyla and ribotypes (Fig. 2). Increasing the number of sequenced clones per sample may have revealed additional rare ribotypes; however, the variance among colonies was not based upon the presence of rare ribotypes. Even when singletons and doubletons (Table 1) were excluded from the analysis, the variances of shared ribotypes among the FST samples remained high.
In contrast to the geographical origin of the FST samples, storage of FST samples in ethanol for several years prior to extraction of the bacterial DNA did have an effect on the measured bacteria community parameters. Ethanol storage reduced ribotype richness, but increased bacterial diversity. This apparent contradiction is explained by the fact that the Bacteroides ribotype, which is predominant in fresh samples, masks the presence of rare species. High number of singletons (i.e., ribotypes represented by only one clone) indicate that there may be a considerable number of ribotypes in the fresh samples that remained undetected. The abundance of Bacteroidetes was reduced in EtOH stored samples and thus rare species were more likely to be detected in the clone library, which increased the bacterial diversity in EtOH stored samples relative to fresh samples.
The bacterial phyla that persisted predominantly in the EtOH-stored FST samples (Firmicutes, Actinobacteria) were also found in bacterial community studies of other long-term preserved specimens. In a study of ancient bacteria sampled in permafrost of different ages (0-8.1 MYA), DNA of Actinobacteria were found to be the most persistent, followed by the DNA of the Firmicutes classes Bacillalaceae and Clostridiaceae [82]. The proportion of gram-positive bacteria increased approximately 50% in the permafrost samples 5-30 kyr of age compared to fresh permafrost samples; in the 300-600 kyr permafrost samples the whole bacterial community consisted of gram positive bacteria [82]. In the intestines of freeze-dried human mummies, Clostridia were dominant and Bacteroidetes, which were abundant in the intestines of living humans [83], were absent. Gram-negative bacteria, which made up ~40% of the bacterial community in the intestine of living humans, dropped to ~20% in the intestine of a 90 year-old mummy and were almost absent in a 3500 year-old Iceman mummy [83]. This was comparable to our study, in which the proportion of Bacteroidetes and other gram-negative bacteria decreased substantially in the FST samples stored long-term in ethanol. The higher persistence of the gram-positive bacterial DNA may be attributed to the multiple layers of peptidoglycan in their cell walls [84], which can protect the DNA much better from the degradation than the thin peptidoglycan and lipopolysaccharide layers in the cell wall of the gram-negative bacteria [85].
CONCLUSION
Data from this study using multiple FST colonies from the native and introduced range increased the number of ribotypes known to inhabit the gut of FST four-fold (213 ribotypes). The enemy release hypothesis (pertaining specifically to the release from pathogens) could not be invoked to explain invasion success of FST since no pathogens were found among the bacterial gut community in the native and introduced populations of FST. Ribotype richness, bacterial diversity, and proportions of detected phyla were influenced by storage conditions of the FST samples, but not by their geographic origin. Invasion of new habitats did not have any detectable impact on the composition of the bacterial gut symbionts. Apparently, the tight co-evolutionary link between termites and their gut flora maintains a certain association of species.
ACKNOWLEDGEMENTS
We thank Drs L. Foil and A. Sethi for providing valuable comments on an earlier draft of this manuscript. Termite samples from Louisiana were provided by the New Orleans Mosquito Termite Control Board and USDA-ARS-SRRC. Samples from China were provided by J. Kenneth Grace (University of Hawaii at Manoa) and Changlu Wang (Rutgers University). This study was funded by the Louisiana Board of Regents Research Competitiveness Subprogram ((LEQSF(2004-2007)-RD-A-01) to CH and the National Science Foundation to MB (DEB-0072741). Approved for publication by the Director, Louisiana Agricultural Experiment Station, as Manuscript No. 2009-234-3833.

