All published articles of this journal are available on ScienceDirect.

Assignment of Reference 5’-end 16S rDNA Sequences and Species-Specific Sequence Polymorphisms Improves Species Identification of Nocardia
Authors Info & Affiliations
Abstract
16S rDNA sequence analysis is the most accurate method for definitive species identification of nocardiae. However, conflicting results can be found due to sequence errors in gene databases. This study tested the feasibility of species identification of Nocardia by partial (5’-end 606-bp) 16S rDNA sequencing, based on sequence comparison with “reference” sequences of well-annotated strains. This new approach was evaluated using 96 American Type Culture Collection (n=6), and clinical (n=90) Nocardia isolates. Nucleotide sequence-based polymorphisms within species were indicative of “sequence types” for that species. Sequences were compared with those in the GenBank, Bioinformatics Bacteria Identification and Ribosomal Database Project databases. Compared with the reference sequence set, all 96 isolates were correctly identified using the criterion of ≥99% sequence similarity. Seventy-eight (81.3%) were speciated by database comparison; alignment with reference sequences resolved the identity of 14 (15%) isolates whose sequences yielded 100% similarity to sequences in GenBank under >1 species designation. Of 90 clinical isolates, the commonest species was Nocardia nova (33.3%) followed by Nocardia cyriacigeorgica (26.7%). Recently-described or uncommon species included Nocardia veterana (4.4%), Nocarida bejingensis (2.2%) and, Nocardia abscessus and Nocardia arthriditis (each n=1). Nocardia asteroides sensu stricto was rare (n=1). There were nine sequence types of N. nova, three of Nocardia brasiliensis with two each of N. cyriacigeorgica and Nocardia farcinica. Thirteen novel sequences were identified. Alignment of sequences with reference sequences facilitated species identification of Nocardia and allowed delineation of sequence types within species, suggesting that such a barcoding approach can be clinically useful for identification of bacteria.
INTRODUCTION
Nocardia species cause a range of infections including localised lung and skin infections, and disseminated disease. Speciation of clinical isolates is important to characterise associated disease manifestations, predict antimicrobial susceptibility and identify differences in epidemiology [1]. Since standard phenotypic identification methods are time-consuming and often imprecise [2, 3], nucleic acid-amplification tools targeting conserved gene regions have been developed to facilitate accurate species determination.
Of these, 16S rDNA sequence analysis is the most frequently-used method for definitive species identification of nocardiae [2, 4-6]. Polymorphisms within the 65-kDa heat shock protein gene (hsp65) target are also reported to enable speciation [7, 8]. These sequence-based identification methods have led to substantial species re-assignment within the genus, especially among “Nocardia asteroides” isolates. Over 80 species have now been described of which at least 33 have been implicated in human disease (http://www. ncbi.nlm.nih.gov/Taxonomy/; http://www.bacterio.cict.fr/n/ nocardia.html; [2]).
Numerous Nocardia 16S rDNA sequences have thus been deposited in public sequence databases; however, a substantial proportion of sequences, for example in GenBank, represents misidentified isolates or contains significant errors [9, 10]. Imprecise species identification may also result from the presence of multiple, but different, copies of 16S rDNA in certain Nocardia spp. such as Nocardia nova [11]. Further, sequence-based analyses are complicated by the lack of consensus regarding the degree of sequence similarity required for species definition of Nocardia [12].
To improve sequence-based species identification, there has been strong impetus to develop libraries of DNA sequences in order to designate, or link, standardised sequences, including nucleotide polymorphisms within these sequences, with a particular species; this process requires the establishment of such “reference sequences” or “DNA barcodes” for species identification and for the recognition of intraspecies sequence polymorphisms or “sequence types” [13, 14]. Such an approach has not yet been applied to the identification of Nocardia. Since the few molecular analyses of Nocardia culture collections have reported significant species misidentification using phenotypic methods [5, 15], we re-examined the species identity of 96 Nocardia isolates in our collection using partial (5’-end 606-bp) 16S rDNA sequencing and the assignment of sequence types. The accuracy of three publicly-available gene databases for species identification was compared.
MATERIALS AND METHODS
Nocardia Organisms
Ninety-six Nocardia isolates were studied (supplementary Table S1 ; Table 1). These comprised six American Type Culture Collection (ATCC; Rockville, MD) strains (N. asteroides ATCC 19247T, Nocardia farcinica ATCC 3308, N. farcinica ATCC 3318T, N. nova ATCC 33726T, Nocardia otitidiscaviarum ATCC 14629T and Nocardia paucivorans ATCC BAA-278T) and 90 clinical isolates (from the Clinical Mycology Laboratory, Centre for Infectious Diseases and Microbiology, Westmead Hospital, Sydney, Australia). Clinical isolates were cultured from separate patients from 1997-2005. All isolates were speciated using standard phenotypic methods and antibiotic susceptibility profiles [3] and, by 16S rDNA sequencing. Isolates were cultured aerobically in brain heart infusion broth (Amyl Media, Dandenong, Australia) for 3-15 days at 37°C.
Species Distribution of 96 Nocardia Isolates by Sequence-Based Alignment of 606-bp 16S rDNA Fragments with Reference Sequences and BLASTn Against Database Sequences
| Species identification (Alignment with Reference Sequences) | No. Isolates/No. Matched by Phenotypic Identification | No. Species with ≥99% Sequence Match (BLASTn) | GenBank Range % Similarity/No. Matched Sequences1 | BIBI Range % Similarity/No. Matched Sequences1 | RDP-II Range % Similarity/No. Matched Sequences1 |
|---|---|---|---|---|---|
| N. asteroides sensu stricto | 2/2 | 1 | 99.0-100/3 | 100/3 | 100/2 |
| N. abscessus | 1/0 | 32 | 100/15 | 100/20 | 100/13 |
| N. aobensis | 1/0 | 1 | 99.8/4 | 99.8/4 | 98.8/4 |
| N. arthritidis | 1/0 | 13 | 99.6/13 | 99.6/13 | 0 |
| N. beijingensis | 2/0 | 1 | 100/13 | 100/18 | 98.1/13 |
| N. brasiliensis | 3/3 | 1 | 100/2 | 100/4 | 100/2 |
| N. cyriacigeorgica | 24/0 | 1 | 99.8-100/35 | 99.8-100/21 | 99.1-100/15 |
| N. farcinica | 13/10 | 24 | 99.8-100/14 | 100/12 | 100/8 |
| N. nova | 31/28 | 1 | 99.3-100/5 | 99.6-100/5 | 99.0-100/4 |
| N. otitidiscaviarum | 5/4 | 1 | 100/5 | 100/5 | 100/4 |
| N. paucivorans | 7/1 | 1 | 100/1 | 100/1 | 100/1 |
| N. transvalensis | 1/0 | 1 | 99.7/3 | 99.6/5 | 98.8/2 |
| N. veterana | 4/0 | 1 | 100/5 | 100/8 | 100/4 |
| N. vinacea | 1/0 | 1 | 100/3 | 100/3 | 100/2 |
1 Refers to the number strains in the database with a sequence similarity of ≥ 99% to that of the query sequence.
2 Refers to 100% sequence similarity with N. asteroides, N. asiatica and N. abscessus sequences.
3 99.6% sequence similarity to a single N. bejingensis sequence (GenBank accession no. AY756543) but <99% sequence similarity to other N. bejingensis strains. Since the sequence demonstrated 99.0% similarity to the reference N. arthriditis sequence, it was assigned as such.
4 Refers to 100% sequence similarity with N. farcinica sequences as well as with sequence of N. otitidiscavarium strain DSM 43242T (GenBank accession no. X80611).
Reference 5’ end 606-bp 16S rDNA Sequences of 45 Nocardia Species1 used in the Present Study
| Strain no.1,2 | Species Identification | GenBank Accession no.3 |
|---|---|---|
| DSM 44432T | N. abscessus | AY544980 |
| DSM 44491T | N. africana | AY756540 |
| IFM 0137 | N. aobensis | AB126875 (bp positions 40-645) |
| DSM 44729T | N. araoensis | AY903623 |
| DSM 44731T | N. arthritidis | AY903619 |
| DSM 44668T | N. asiatica | AY903617 |
| ATCC 19247T | N. asteroids | AY756541 |
| ATCC 49872 | N. asteroides type IV | AY756542 |
| JCM 10666T | N. beijingensis | DQ6599014 (bp positions 14-619) |
| JCM 10666T | N. beijingensis | AY756543 |
| ATCC 19296T | N. brasiliensis | AY756544 |
| DSM 43024T | N. brevicatena | AY756545 |
| DSM 43397T | N. carnea | AY756546 |
| DSM 44546T | N. cerradoensis | AY756547 |
| ATCC 700418T | N. crassostreae | AY756548 |
| DSM 44490T | N. cummidelens | AY756549 |
| DSM 44484T | N. cyriacigeorgica | AY7565505 |
| ATCC 14759 | N. asteroides type VI | DQ2238625 |
| DSM 44890T | N. elegans | DQ659905 (bp positions 1-603)6 |
| DSM 43665T | N. farcinica | AY756551 |
| JCM 3332T | N. flavorosea | AY756552 |
| DSM 44489T | N. fluminea | AY756553 |
| DSM 44732T | N. higoensis | AY903620 |
| DSM 44496T | N. ignorata | AY756554 |
| DSM 44667T | N. inohanensis | AY903611 |
| CIP 108295T | N. mexicana | AY903610 |
| DSM 44717T | N. neocaledoniensis | AY903614 |
| DSM 44670T | N. niigatensis | AY903615 |
| CIP 104777T | N. nova | AY756555 |
| ATCC 14629T | N. otitidiscaviarum | AY756556 |
| DSM 44386T | N. paucivorans | AY756557 |
| DSM 44730T | N. pneumonia | AY903622 |
| DSM 44290T | N. pseudobrasiliensis | AY756558 |
| DSM 43406T | N. pseudovaccinii | AY756559 |
| DSM 44599T | N. puris | AY903618 |
| JCM 4826T | N. salmonicida | AY756560 |
| DSM 44129T | N. seriolae | AY756561 |
| DSM 44733T | N. shimofusensis | AY903621 |
| DSM 44488T | N. soli | AY756562 |
| DSM 44704T | N. tenerifensis | AY903613 |
| DSM 44765T | N. testacea | AY903612 |
| DSM 43405T | N. transvalensis | AY756563 |
| JCM 3224T | N. uniformis | AY756564 |
| ATCC 11092T | N. vaccinii | AY756565 |
| DSM 44445T | N. veteran | AY756566 |
| JCM 10988T | N. vinacea | AY756567 |
| DSM 44669T | N. yamanashiensis | AY903616 |
1 All species designations were cross-checked against two websites - http://www.ncbi.nlm.nih.gov/Taxonomy/ and http://www.bacterio.cict.fr/n/nocardia.html. Abbreviations: ATCC, American Type Culture Collection; CIP, Collection Institut Pasteur, France; DSM, Deutsche Sammling von Mikroorganismen und Zellkulturen GmbH, Germany; JCM, Japan Collection of Microorganisms, Wako-Shi, Japan. Adapted from [7, 17, 18].
2 T refers to the previously-designated current type strains for the particular species.
3 Unless otherwise specified refers to first 1-606 bp of the 16S rDNA sequence from the 5’end.
4 Different sequence results obtained for N. beijingensis JCM 10666T in two separate studies. The sequence with GenBank accession no. DQ659901 is chosen as the reference sequence.
5 The sequence of N. cyriacigeorgica strain DSM 44490T (GenBank accession no. AY756550) is identical to the sequence of N. asteroides ATCC 14759 (accession no. DQ223862).
6 The sequence is based on a 603-bp 16S rDNA fragment.
Partial (5’-end 606-bp) 16S rDNA Sequence Polymorphisms in 10 Nocardia Species
| Strain Identification no. | Identification Based on Comparison with Reference Sequence | % Similarity to Reference Sequence | Site of Nucleotide Polymorphisms (bp Position: Reference Sequence → Isolate Sequence) | 100% Similarity to Another GenBank Sequences Rather Than Reference Sequences (13 Novel Sequence GenBank Accession no.) |
|---|---|---|---|---|
| 04-303-0576 | N. aobensis | 99.67 | 137 G→A | novel sequence1 (FJ172101) |
| 01-320-2714 | N. arthritidis | 99.0 | 132 C→T, 240 G→A, 251 C→T, 341 C→G, 566 A→G, 588 G→T | novel sequence1 (FJ172102) |
| 02-071-3627 | N. asteroides | 99.0 | 133-135 TTC→ACA, 148-150 GAG→TGT | novel sequence1 (FJ172103) |
| 00-194-3516 | N. brasiliensis | 99.84 | 203 T→C | Z36935 |
| 03-273-2825 | N. brasiliensis | 99.67 | 203 T→C, 328 G→A | AY245543 |
| 99-167-2395 | N. brasiliensis | 99.67 | 203 T→C, 328 G→A | AY245543 |
| 00-159-1584 | N. cyriacigeorgica | 99.84 | 576 G→A | novel sequence1 (FJ172112) |
| 04-181-3939 | N. cyriacigeorgica | 99.84 | 576 G→A | |
| 05-111-2308 | N. cyriacigeorgica | 99.84 | 576 G→A | |
| 01-109-2248 | N. farcinica | 99.84 | 67 A→A/G2 | novel sequence1 (FJ172117) |
| 05-053-4454 | N. nova | 99.84 | 86 C→ C/T2 | novel sequence1 (FJ172126) |
| 02-352-3316 | N. nova | 99.84 | 136 G→A/G2 | novel sequence1 (FJ172120) |
| 00-130-2170 | N. nova | 99.84 | 260 A→A/G2 | novel sequence1 (FJ172121) |
| 04-110-3287 | N. nova | 99.67 | 86 C→C/T2, 136 G→A/G2 | novel sequence1 (FJ172125) |
| 00-056-3529 | N. nova | 99.67 | 86 C→T, 136 G→A | AF430030 |
| 01-067-1349 | N. nova | 99.67 | 86 C→T, 136 G→A | |
| 01-097-0996 | N. nova | 99.67 | 86 C→T, 136 G→A | |
| 02-199-2723 | N. nova | 99.67 | 86 C→T, 136 G→A | |
| 00-025-0538 | N. nova | 99.67 | 135 G→T, 148 T→G | AF430032 |
| 00-314-1789 | N. nova | 99.67 | 135 G→T, 148 T→G | |
| 04-150-0614 | N. nova | 99.67 | 135 G→T, 148 T→G | |
| 01-066-1903 | N. nova | 99.51 | 86 C→T, 136 G→A, 578 T→G | novel sequence1 (FJ172122) |
| 02-021-0419 | N. nova | 99.51 | 135 G→T, 148 T→G, 328 A→G | novel sequence1 (FJ172119) |
| 01-114-2816 | N. otitidiscaviarum | 99.85 | 133 A→G | novel sequence1 (FJ172127) |
| ATCC BAQ-278 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | AF179865 |
| 03-141-3073 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 03-185-3304 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 03-240-2758 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 05-200-1797 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 97-114-0609 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 97-16298 | N. paucivorans | 99.67 | 33 C-ins3, 37 G-ins3 | |
| 03-310-2776 | N. transvalensis | 99.67 | 402, 403 AG→CA | novel sequence1 (FJ172131) |
1 Sequences of isolates without a match with GenBank sequences are novel sequences.
2 At the specified bp position, both nucleotides were present due to different multiple copies of the 16S rRNA gene.
3 Refers to insertion of the specified base.
DNA Extraction
Cells from 2 ml-brain heart infusion broth cultures of Nocardia in late logarithmic phase were harvested by centrifugation at 14,000 X g for 10 min. The supernatant was removed and the pellet suspended in 150 μl of digestion buffer (10 mM Tris-HCl [pH 8.0], 0.45% Triton X-100 and 0.45% Tween 20). Bacterial suspensions were then heated for 10 min at 100°C to lyse the cells, followed by cooling at -20°C for 1 h. Cell lysates were centrifuged at 14,000 X g for 5 min to pellet the cell debris. Supernatants containing DNA were diluted in 350 μl TE buffer (5mM Tris HCl, 0.5 mM EDTA) and centrifuged for 2 min to remove cell debris. DNA was quantitated using a spectrophotometer and stored at –20°C until required.
PCR Amplification and Sequencing of the 16S rDNA
The 5’-end 606-bp fragment of the 16S rDNA gene was amplified using the universal bacterial primers 16S-27f0 (5’ to 3’: 1 TTA GAG TTT TGA TCM TGG CTC 21) and 16S-907r (5’ to 3’: 986 CCG TCA ATT CMT TRA GTT T 877) [16]. Each PCR reaction contained 5 µl template DNA, 0.25 µl (50 pmol/µl) each of forward primer and reverse primer, 1.25 µl dNTPs (2.5 mM of each dNTP: Roche Diagnostics, Mannheim, Germany), 2.5 µl 10x PCR buffer (Qiagen, Donacaster, Victoria), 0.1 µl HotStar Taq polymerase (5 U/µl) and water to a 25 µl final volume. Amplification was performed in a Mastercycler gradient thermocycler (Eppendorf; Netheler-Hinz GmbH, Germany). The cycling conditions were: 95°C for 15 min followed by 35 cycles of 94°C for 30 s, 55°C for 30 s, 72°C for 90 s with a final extension step at 72°C for 10 min.
PCR products were purified (PCR Product Pre-sequencing Kit; USB Corporation, Cleveland, OH) and sequenced using the BigDye Terminator version 3.1 cycle sequencing kit (ABI PRISM 3100 genetic analyser; Applied Biosystems, Foster City, CA) and the primer 16S-27f (5’ to 3’: 3 AGA GTT TTG ATC MTG GCT CAA G 23) [16]. Each sequence was manually aligned and analysed to ensure high quality sequence data. Where the sequence of an isolate differed from the GenBank “reference” sequence for that species (see Results and Table 2), [7, 17, 18] or for novel sequences, additional primers (16S-27f0 and 16S-907r) were used to confirm the result of the sequence.
16S rDNA Sequence Analysis
For each isolate, the amplified 5’-606 bp 16S rDNA fragment was examined using the BioManager facility (ANGIS, Sydney; http://biomanager.angis.org.au/). Consensus sequences were constructed from alignments of sequence data using ClustalW [19] after careful examination of each electrophoregram trace representation of data. Sequence data were queried against archived sequences in the GenBank (BLASTn 2.2.10; http://www.ncbi.nlm.nih.gov), Bioinformatics Bacteria Identification (BIBI) version 0.2 (http:// umr5558-sud-str1.univ-lyon1.fr/lebibi/lebibi.cgi; [20]) and Ribosomal Database Project-II (RDP-II) version 9.54 (http://rdp.cme.msu.edu/index.jsp; [21]) databases.
A list of the closest sequence matches was generated from the database comparisons with pair-wise distance scores indicating the percent similarity between the unknown (query) sequence and database sequences. Only consensus sequences with a minimum length of 606-bp were analysed. A percent similarity (or identity) score of ≥ 99% [4, 5] was used as the criterion to classify an isolate to species level whilst a 97 to 98.9% similarity score identified an isolate as belonging to the genus Nocardia but to a different species [9, 10].
Nucleotide Sequence Accession Number
Thirty-four 606 bp partial 16S rDNA sequences including 13 novel Nocardia sequences generated in the study were deposited in GenBank with the following accession numbers (also see Table 3): FJ172101 through to FJ172134.
RESULTS
Establishment of a Reference Set of 16S rDNA Sequences
For this study, the 606-bp 16S rDNA sequences of 43 well-characterised isolates representing 43 taxonomically-authenticated Nocardia species (http://www.bacterio.cict.fr/n/nocardia.html) were chosen as the reference sequences for that species; these isolates were previously-characterised by both 16S rRNA and hsp65 gene analyses [7, 8]. The sequences of two additional isolates, Nocardia elegans DSM 44890T and Nocardia aobensis IFM 0137 [17, 18], were included in the reference sequence set (Table 2). Examination of the N. asteroides ATCC 14759 sequence (N. asteroides type VI, GenBank accession no. DQ223862) found it to be identical to that of Nocardia cyriacigeorgica DSM 44484T (GenBank accession no. AY756550). The sequence of Nocardia beijingensis JCM 10666T (GenBank accession no. AY756543) reported in one study [6, 7] was 98.5% similar to that of this same strain (GenBank accession no. DQ659901) described in a separate study [17]. We assigned the sequence corresponding to accession no. DQ659901 as the reference sequence for N. beijingensis since this yielded the higher similarity (100%) to multiple N. beijingensis sequences in the GenBank, BIBI and RDP-II databases.
This collection of partial 16S rDNA sequences representing 45 Nocardia species formed the basis for re-evaluating the species identity of the study isolates and was important in the validation of the sequence analyses. Strains fulfilling the criterion for a species (≥99% sequence similarity) but which demonstrated sequence polymorphisms compared to the reference sequence for that species were considered as separate sequence types for the species.
16S rDNA Sequence-Based Identification of Nocardia Isolates
The details of 96 Nocardia isolates identified by phenotypic methods and partial 16S rDNA sequencing are given in supplementary Table S1. The species distribution of isolates, as determined by sequence comparison with the reference sequence set, and with sequences in the GenBank, BIBI and RDP-II databases, is shown in Table 1.
(a). Identification Based on Comparison with Reference 606-bp 16S rDNA Sequences
Following alignment of sequence data with the reference sequence set, partial 16S rDNA sequencing provided species identification for all 96 isolates using a criterion of ≥99% sequence similarity for species definition; 83 (86.5%) isolates were identified if a criterion of 100% sequence similarity was used.
Of 90 clinical isolates, phenotypic methods correctly identified 42 (46.7%) strains to species level (Table 1; supplementary Table S1). Thirty-four isolates were assigned a phenotypic identification of N. asteroides/N. asteroides complex based on their drug susceptibility pattern but 16S rDNA sequencing recognised them as a number of distinct species e.g. N. cyriacigeorgica,N. farcinica and N. paucivorans among others (Supplementary Table S1). Molecular and phenotypic species identification methods were concordant for 80% (8 of 10), 75% (three of four), 90% (27 of 30) and 100% (all of three) of N. farcinica, N. otitidiscavarium, N. nova and Nocardia brasiliensis clinical isolates, respectively. However, none of the six N. paucivorans isolates were correctly identified by phenotypic methods. All six ATCC strains were assigned by 16S rDNA sequencing to their respective species (Supplementary Table S1).
Discrepant results for isolates are summarised in Supplementary Table S1 (see also Table 1). In particular, only one of 35 phenotypic “N. asteroides/N. asteroides complex” isolates (other than strain ATCC 19247T) had a sequence identical to sequences of N. asteroides sensu stricto (represented by N. asteroides ATCC 19247T); 22 clinical isolates had sequences with 100% similarity to the reference N. cyriacigeorgica sequence. The remaining strains were N. paucivorans (n=3), N. nova (n=2), N. beijingensis (n=2), Nocardia abscessus (n=1), Nocardia arthritidis (n=1), N. farcinica (n=1) and Nocardia veterana (n=1). Other discrepant results included two N. farcinica (phenotypic identification) isolates yielding sequences with 100% similarity to the reference N. cyriacigeorgica sequence and a N. brasiliensis strain with 100% sequence similarity to N. otitidiscavarium.
Eleven isolates (supplementary Table S1) identified as “Nocardia spp.” by phenotypic methods were identified as N. paucivorans (n=3), N. farcinica, N. veterana (each n=2) and N. aobensis, N. nova, Nocardia transvalensis and Nocardia vinacea (each n=1).
b). Species Identification Based on BLASTn Alignments
Isolates were also identified to species level by comparison with database sequences with the following exceptions: firstly, N. farcinica could not be definitively speciated; all 13 strains (100% sequence similarity to the reference N. farcinica sequence) were identified as either N. farcinica or N. otitidiscavarium (Table 1). The three databases contained a sequence corresponding to N. otitidiscavarium strain DSM 43242T (GenBank accession no. X80611). This sequence was indistinguishable from N. farcinica sequences but only had 94.7% similarity to the reference sequence of N. otitidiscavarium (GenBank accession no. AY756556). Secondly, a phenotypic N. asteroides isolate was identified as N. asteroides/N. abscessus/Nocardia asiatica (Table 1). Since its sequence was identical to the reference sequence of N. abscessus, it was assigned as such. The reference 16S rDNA sequences of N. abscessus and N. asiatica (Table 2) differ only by a single nucleotide polymorphism (SNP) at position 527 (“G” for N. abscessus but “C” for N. asiatica). Finally, an isolate (strain 01-320-2714; supplementary Table S1) was identified as N. beijingensis by database comparisons (99.6% sequence similarity to a single N. bejingensis sequence; GenBank accession no. AY756543). However, as it yielded 99% sequence similarity (5-bp difference) to the reference sequence of N. arthriditis, it was assigned as N. arthriditis (Table 1).
Comparison of Species Identification Using GenBank, BIBI and RDP-II Databases
For all isolates, the same species identification result was obtained by comparison of their sequences with those in the GenBank, BIBI and RDP-II databases (Table 1). The distribution of percent similarity scores according to database is shown in Fig (1). Using the criterion of ≥99% sequence similarity for species designation, all 96 isolates were identified by the GenBank and BIBI databases and 91 (96.7%), by the RDP-II system. The length of sequences employed for sequence alignment in the BIBI, RDP-II and GenBank databases ranged from 512-548 bp, 572-590 bp and ≥606 bp, respectively. Perfect matches (100% similarity) were observed for 77%, 79% and 93% of sequence alignments against the GenBank, BIBI and RDP-II databases, respectively (Fig. 1).
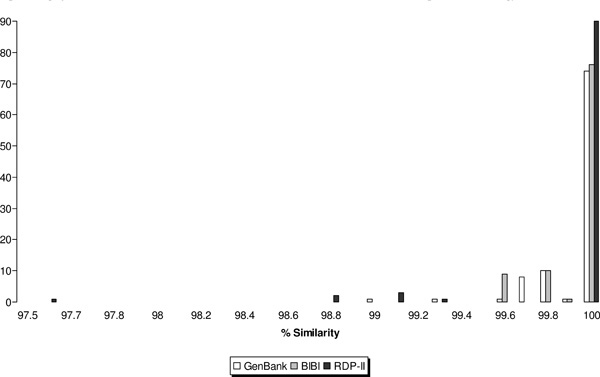
Distribution of similarity scores for partial 16S rDNA sequence-based identification of Nocardia isolates by the GenBank, BIBI and RDP-II databases.
Species Distribution of Clinical Isolates
Fourteen Nocardia species were identified amongst 90 isolates, the most common being N. nova (30 isolates; 33%) followed by N. cyriacigeorgica (n=24; 27%), N. farcinica (n=11; 12%) and N. paucivorans (n=6; 7%). There were four isolates each of N. otitidiscaviarum and N. veterana, three strains of N. brasiliensis, two of N. beijingensis and one each of N. asteroides sensu stricto, N. transvalensis, N. vinacea, N. aobensis, N. arthritidis and N. abscessus.
Intra-Species Variation and Sequence Types of Clinical Isolates
Partial 16S rRNA sequences of N. veterana, N. abscessus, N. beijingensis and N. vinacea isolates were identical to the reference sequence for that species, thus there was a single sequence type. Intraspecies sequence heterogeneity was evident in the remaining 10 species, and varied with species (Table 3). The largest number of sequence types, including that identical to the reference sequence, was noted for N. nova (nine sequence types) followed by N. brasiliensis (three sequence types). N. cyriacigeorgica, N. farcinica, N. asteroides sensu stricto and N. otitidiscavarium exhibited two sequence types. The sequences of three N.cyriacigeorgica isolates (indistinguishable from one another) differed from the reference N. cyriacigeorgica sequence by a SNP at position 576 (substitution of “A” for “G”, see Table 3). There was only one sequence type for N.aobensis, N. transvalensis, N. arthritidis and N. paucivorans but the sequences of these isolates all demonstrated SNPs when compared to the reference sequence for the species (Table 3). Thirteen novel Nocardia sequences were identified for 15 isolates (eight species).
DISCUSSION
Molecular-based identification of Nocardia spp. remains a challenge due to the increasing recognition of new species and changes in taxonomy [22]. Sequence analysis of the 16S rDNA is the current gold standard for identification of Nocardia spp. However few studies have explored the validity of sequence-based identification approaches using collections of phenotypically-characterised clinical isolates [9]. The present study proposes, and has tested the utility of a set of reliable reference Nocardia 16S rDNA sequences, derived from authoritatively–identified organisms [7, 17], as sequence standards for species identification. Further, by identifying and assigning sequence types to represent sequence polymorphisms within a species, the results have identified that such a “barcoding” approach can improve systematic and accurate species identification. DNA barcoding has been designed to provide rapid accurate species identification by using short, standardised gene regions (in this case, the 5’-end 16S rDNA region) as internal species tags [14]. Although it has been found to be effective in speciating eukaryote organisms and some parasites [13, 14, 16], there are few data on its application in the identification of bacterial pathogens.
As such, based on comparison with the reference sequence set, partial 16S rDNA sequencing provided clear species identification of all 96 isolates. In particular, the reference sequence set was useful in the speciation of 11 isolates identified only to genus level by phenotypic methods and assisted in resolving the identity of 14 (15%) clinically relevant isolates whose sequences aligned with 100% similarity to sequences assigned to more than one species in the GenBank, BIBI and RDP-II databases (Table 1). Without the resource of this sequence set, sequencing was unable to assign precise species identification to 13 N. farcinica isolates (Table 1). It is most likely that the N. otitisdiscavarium DSM 43242T sequence (GenBank accession no. X80611) with 100% identity to N. farcinica sequences represents a misidentification, underscoring the importance of adequate stewardship of database sequences. Of note, the reference sequence of N. abscessus and N. asiatica (GenBank accession nos. AY544980 and AY903617, respectively) differ only by a SNP - this low interspecies heterogeneity likely explains the inability to resolve species identification for N. abscessus after alignment with databases sequences (Table 3). N. abscessus (previously N. asteroides antimicrobial susceptibility type I) represents ≈20% of isolates of the former N. asteroides complex; accurate species identification is important as MICs of imipenem, which is commonly used to treat nocardiosis, are high for this species [2]. There are few descriptions of N. asiatica as a pathogen.
Species identification of Nocardia by gene sequence analysis is heavily reliant on the entries in the gene repositories being queried [10, 23]. As noted in the present study, inappropriate and /or obsolete sequence entries are important potential limitations. Further, species identification based on data derived from a single or small numbers of strains representing a species must be interpreted with caution (see also Table 1). As such, the submission of carefully-annotated new sequences is critical to maintaining the accuracy of current gene repositories. Since the BIBI and RDP-II systems contain a larger proportion of shorter 16S rDNA sequences (512-548 bp, 572-590 bp, respectively vs. ≥606 bp in GenBank), this may have resulted in an artificially inflated number of perfect sequence matches (Fig. 1). The validity of comparisons with sequences of different lengths for species identification requires further study.
The approach undertaken in this study has further allowed us to distinguish between closely-related Nocardia species, to identify newly described or uncommon species and to determine the species distribution of clinical Nocardia isolates received in our laboratory. Elsewhere and in Australia, most human infections historically have been attributed to N. asteriodes sensu stricto antimicrobial susceptibility class types I and VI, N. nova, N. brasiliensis and N. farcinica [1, 24]. Our results reconfirm that this generally remains the case. Given the prevalence of N. cyriacigeorgica (previously N. asteroides type VI as a pathogen (this study; [25]), adoption of protocols by microbiology laboratories for its identification is important. Apart from distinguishing N. abscessus from N. asiatica (see above), partial 16S rDNA sequencing was able to differentiate between other closely-related species including N. veterana and N. nova sensu stricto (sequence similarity of 98.1%; [6, 26, 27]). Classified within the N. nova complex, N. veterana is an emerging pathogen capable of causing serious infection [27]. Of note, the re-assignment of all but one clinical “N. asteroides” strains to other taxonomic groups questions the validity of N. asteroides as a separate species.
Importantly, the results identified significant intraspecies sequence polymorphisms within the 16S rDNA for many (10 of 14) Nocardia species or for seven of nine species represented by more than one strain (Table 3), and that such nucleotide heterogeneity differed according to species, being most evident for N. nova (nine sequence types). Although there was no genetic heterogeneity amongst isolates of, for example, N. farcinica, the sequences of the isolates differed from the reference sequence for this species. Thus, if constructing a DNA template or “identification barcode” for identifying Australian N. paucivorans isolates, 16S rDNA position 33 should incorporate an extra C and at position 37, an extra G in relation to the reference sequence (Table 3). In the USA, three “genetic types” of N. cyriacigeorgica with SNPs at positions 448, 1427 and 1480 [25] have been reported; we identified a SNP at position 576 but not at position 448. Thus, comparison of sequence types of isolates from different countries to identify potential clinical and epidemiological associations may be warranted. As noted for other bacteria, substitutions of as little as 1-2 bp may correlate with unique Nocardia phenotypes and clinical significance [5, 28]. Therefore, documentation of sequence polymorphisms within species is relevant to delimiting species or highlighting genetically-distinct groups with levels of sequence divergence that are either suggestive or exclusive of species status.
Finally, 13 novel sequences from eight Nocardia species were identified and some of them may merit description as new species (Table 3). Species designation, however, is confounded by the lack of a consensus criterion for species definition based on percent similarity scores; isolates of distinct species of Nocardia have been reported to exhibit as much as 99.8% sequence similarity [12].
CONCLUSION
Partial 16S rDNA sequencing is a viable alternative to full-length sequencing for species identification of Nocardia in a diagnostic laboratory. The present approach encompassed comparison of the sequence of interest with a library of reference sequences and the assigning of sequence types to represent sequence polymorphisms within species, based on sequence similarity to the reference sequence for that species; unambiguous species identification was obtained for all study isolates. As affirmed in the present study, errors in sequence entries remain important potential limitations of public gene repositories [6, 10, 23]. The results of the study suggest that a barcoding approach [29, 30] can assist with species identification of clinically relevant Nocardia. Further studies are warranted to explore its wider application in improving species differentiation and unravelling sequence data for phylogenetically-unresolved groups of bacteria.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Web site along with the published article.
ACKNOWLEDGMENTS
We thank Ms. Maryann Pincevic for her assistance in performing the 16S rDNA sequencing and Ms. Ping Zhu for help with the Figure preparation.

