All published articles of this journal are available on ScienceDirect.

Presence of Actinobacterial and Fungal Communities in Clean and Petroleum Hydrocarbon Contaminated Subsurface Soil
Authors Info & Affiliations
Abstract
Relatively little is known about the microbial communities adapted to soil environments contaminated with aged complex hydrocarbon mixtures, especially in the subsurface soil layers. In this work we studied the microbial communities in two different soil profiles down to the depth of 7 m which originated from a 30-year-old site contaminated with petroleum hydrocarbons (PHCs) and from a clean site next to the contaminated site. The concentration of oxygen in the contaminated soil profile was strongly reduced in soil layers below 1 m depth but not in the clean soil profile. Total microbial biomass and community composition was analyzed by phospholipid fatty acid (PLFA) measurements. The diversity of fungi and actinobacteria was investigated more in detail by construction of rDNA-based clone libraries. The results revealed that there was a significant and diverse microbial community in subsoils at depth below 2 m, also in conditions where oxygen was limiting. The diversity of actinobacteria was different in the two soil profiles; the contaminated soil profile was dominated by Mycobacterium -related sequences whereas sequences from the clean soil samples were related to other, generally uncultured organisms, some of which may represent two new subclasses of actinobacteria. One dominating fungal sequence which matched with the ascomycotes Acremonium sp. and Paecilomyces sp. was identified both in clean and in contaminated soil profiles. Thus, although the relative amounts of fungi and actinobacteria in these microbial communities were highest in the upper soil layers, many representatives from these groups were found in hydrocarbon contaminated subsoils even under oxygen limited conditions.
INTRODUCTION
Soil microbial communities are among the most complex and diverse assemblages in the biosphere and they play an important role in all the ecosystem services provided by soils. Therefore microbial processes in soils have been frequently studied especially in the uppermost soil layers. In contrast, studies of subsurface soil environments have often not adequately characterized microbial community composition and diversity partly because of the cost and difficulty in obtaining large numbers of samples. Therefore, it is not clear if the subsurface microbial communities are closely linked to the surface soil microbes or if they represent a distinct microbial community [1]. The role of microbial communities and especially their activities in subsurface environments is receiving increased attention due to their importance to ecosystem function, agriculture and environmental management [2, 3]. The bioremediation processes, where micro-organisms degrade hazardous compounds like petroleum hydrocarbons (PHCs) to non-hazardous inorganic compounds, is an example of environmental services provided by soils. The hazardous compounds will in most cases not be restricted to the upper soil layer but will eventually percolate to the subsurface [4], where they are a potential threat to the quality of groundwater resources.
In Finland 200 new cases of environmental contamination due to oil leakage are reported annually. Bioremediation processes in surface soils are often efficient and can be monitored relatively easily. Less is known about the microbial populations in soil environments contaminated with complex hydrocarbon mixtures, especially in the subsurface soil layers. It is likely, that the degrading micro-organisms and the degradation processes in subsurface soils differ from the surface soils because of differences in the chemical and physical environment (e.g. organic matter content and gas composition). Traditionally bioremediation studies have focused on fast growing and nutrient-demanding bacterial groups such as Pseudomonas. It is possible however, that these traits are not very useful in subsurface soils where nutrients are scarce and the environmental conditions are harsh. Therefore the use of such organisms for bioremediation purposes may often have been unsuccessful due to limited knowledge of prevailing in situ conditions and microbial requirements during the bioremediation process. Moreover, the microbial community composition varies between soil types, and different populations of degraders therefore become dominant in different soils [5].
In subsurface soils, the substrate consumption leads with time to mass transfer-limited conditions and soil populations are most likely not growing, except for transient growth on decaying cells [6, 7]. These conditions are likely to favor microbes with low growth rates and long-term survival characteristics like low energy demands as well as investment of energy in maintenance. Gram positive bacteria and fungi are typical representatives of this group of micro-organisms. The Gram positive actinobacteria are aerobes or microaerophils, but it is not known if they posses possible anaerobic degradation properties. The actinobacteria have been encountered in late succession stages of soil [8] and they have also been reported to increase with increasing soil depth [9]. Members of the actinobacteria have been reported to aerobically and microaerobically degrade hydrocarbons in many soils [5], including alpine [10] and polar soils [11]. Most fungi are obligate aerobes, but many yeasts and several mycelial fungi are facultative anaerobes, being able to ferment sugars or to use nitrate or sulfate as alternative electron acceptors. However, there is hardly any information on the anaerobic degradation of contaminants by fungi. Some fungi are able to aerobically degrade a wide range of PHCs and are thought to play an important role in the primary attack of poorly available pollutants making them more susceptible to bacterial degradation [12]. Under some conditions fungi may even be more effective degraders than bacteria [13], since the initial attack on high molecular weight hydrocarbons by fungal exoenzymes may give them an advantage compared to bacteria in soil [6]. Therefore the role of fungi in degradation of PHCs may be important. Until now studies on fungi and actinobacteria that degrade PHCs in subsoil are scarce. The reason that these groups of microbes have not received the attention they deserve is that they are slow growing and often demanding to culture by current culturing techniques. With the use of molecular tools, which take advantage of the species specific nucleic acids sequences, the microbial communities in different environments can be described without the need of culturing them in laboratory conditions. Recent studies on microbial community composition using cloning and sequencing of PCR amplified bacterial 16S rDNA have shown that actinobacteria are commonly found in small amounts in anoxic contaminated environments. Dojka and coworkers [14] found that actinobacteria constituted 0.5 % of the members of the bacterial domain in a hydrocarbon and solvent contaminated anoxic aquifer. Kasai and coworkers [15] found a 2 % proportion of all bacteria to belong to actinobacteria in an anoxic soil. To our knowledge, no studies of contaminated subsurface so far have included fungi in their community analyses.
In the present study we estimated the biomass and composition of micro-organisms in two different soil profiles down to 7 m depth originating from an aged contaminated soil profile that was contaminated with PHCs and a clean soil profile next to the contaminated site. Special interest was paid to investigating the fungal and actinobacterial communities since little is known about the role of these groups for in situ remediation of PHC contaminated subsurface soils.
MATERIAL AND METHODS
Soil Samples
Soil samples were taken from a closed landfill in Southern Finland where PHC containing wastes have been dumped 30 years ago. The study area is known to be heavily contaminated with lightweight fuel and lubrication oils in both the saturated and unsaturated zone and partly with heavy metals in the top soil [16]. Samples were taken from two sampling points using a medium size drilling rig applying the core sampler technique. The soil profile from the highly contaminated point was divided into 0.5 m subsamples and the soil profile from a corresponding clean reference point was divided into 1 m subsamples. The samples were sieved at the site with a 8 mm sieve, stored in glass jars and frozen until analysis. The dry weights, ignitions losses and mineral soil concentrations were determined according to [16]. The oxygen levels at the sampling points were measured from gas monitoring wells situated a few meters from the sampling points.
PLFA
Phospholipid fatty acids (PLFAs) were extracted from 45 g of soil and analysed on a Perkin Elmer AutoSystem XL gas chromatograph using the software TurboChrom (version 6.1.1) as described previously [3]. Individual fatty acid methyl esters were identified using a pre-established data base [17] and quantified by means of an internal standard (nonadecanoate methyl ester 19:0).
PLFAs representing bacterial biomasses were i15:0, 15.0, i16:0, 16:1ω7t, i17:0, a17:0, 17:0, cy17:0, 18:1ω7 and cy19:0 [18]. Guilds or single PLFAs representative of specific taxonomical groups were treated as biomass estimates using the fatty acids 10Me17:0 and 10Me18:0 indicative of actinomycetes, 18:2ω6 and 18:2ω9 indicative of fungi, cy17:0, cy19:0 and 18:1ω7 for Gram negative bacteria, i15:0, a15:0, i16:0, i17:0 and a17:0 for Gram positive bacteria, and 16:1ω5 found in arbuscular mycorrhizal fungi and certain Gram negative bacteria of the Cytophaga/Flexibacter-group [19].
Principal component analyses (PCA) on the PLFA data were performed using standardized mol% values. The tests were carried out using correlation matrices on the MatLab software (version 6, Mathworks, Inc., Natick, Ma, USA) and one way ANOVA as well as Tukey's-b Post Hoc tests were performed by SPSS version 11.5.1. (SPSS Inc., Chicago, IL, USA).
DNA Extraction, PCR and DGGE
DNA was extracted from soil samples with the PowerSoil DNA isolation kit (Mo Bio Laboratories, Inc.) using 0.25-0.3 g soil. DNA concentrations were determined by the Quant-iT™ PicoGreen ® dsDNA Assay kit. Actinobacteria-specific 16S rDNA primers S-C-Act-235-a-S-20 and S-C-Act-878-a-A-19 were used [20]. For DGGE analysis the following GC-clamp was added to the S-C-Act-235-a-S-20 primer: 5'-CGC CCG CCG CGC CCC GCG CCC GGC CCG CCG CCC CCG CCC C -3' [21]. The final concentrations in the 50 µl PCR reactions containing 1 µl template were 1 x Taq buffer containing (NH4)2SO4 (MBI Fermentas), 500 μg ml-1 BSA (New England Biolabs), 2 mM MgCl2 (MBI Fermentas), 200 μM of each nucleotide (MBI Fermentas), 200 nM of each primer, 0.5 U Taq polymerase (MBI Fermentas). After a first denaturing step (95°C 4 min) amplification for DGGE analysis was performed in a PTC-200 Thermal Cycler (MJ Research) for 30 cycles in the following way; 95°C, 45 s; 59°C, 45 s; 72°C, 1 min and a final elongation step (72 °C, 5 min). The PCR product was 640 bp long. The annealing temperature was 66 °C for cloning.
For fungal PCR reactions nested primers were used. In the first PCR reaction the 18S rDNA specific fungal primers nu-SSU-0817-5’ and nu-SSU-1536-3’ were used [22]. In the second PCR reaction nu-SSU-0817-5’ and nu-SSU-1196-3’ were used resulting in a 420 bp long product. For DGGE analysis the following GC clamp was added to the nu-SSU-1196-3’ primer: 5'-CGC CCG GGG CGC GCC CCG GGC GGG GCG GGG GCA CGG GGG G-3' [23]. The final concentrations in the 50 µl PCR reactions containing 1 µl template were 1 x Taq buffer containing KCl (MBI Fermentas), 500 μg ml-1 BSA (New England Biolabs), 2.5 mM MgCl2 (MBI Fermentas), 250 μM of each nucleotide (MBI Fermentas), 400 nM of each primer, 0.5 U Taq polymerase (MBI Fermentas) and 2 % formamide (BDH Laboratory Supplies). After a first denaturing step (94°C, 5 min) amplification was performed in a PTC-200 Thermal Cycler (MJ Research) for 35 cycles in the following way; 94°C, 45 s; 50°C, 45 s; 72°C, 1 min and a final elongation step (72°C, 5 min).
Denaturing gradient gel electrophoresis (DGGE) was performed using the DcodeTM Universal Mutation Detection System (BIO-Rad, Hercules, CA, USA), using 6 % (w/v) polyacryleamide gels in 0.5 x TAE (20 mM Tris; 10 mM acetate, 0.5 mM EDTA). The denaturing gradient was 35-65 % for actinobacterial DNAs and 15-50 % for fungal DNAs. On top of the gradient gels a stacking gel without denaturant was added. A volume of 2-25 μl of samples were used. Electrophoresis was run for at 150 V for 10 min followed by 70 V for 18 h. Gels were stained with SYBR Green (Cambrex) for 30 min and the gels were photographed using the Bio-Rad Gel Doc 2000 equipment and the Quantity One -software (Bio-Rad, Hercules, CA, USA). Each band in the DNA profile was assumed to represent one microbial type.
Genomic Libraries
Samples from both clean and contaminated soil profiles corresponding the soil layers with highest PHC concentrations in the contaminated profile were selected for more detailed studies of the microbial species composition using cloning and sequencing. Cloning was performed from one parallel soil sample for Actinobacteria and two parallel samples for fungi. Cloning was performed using the pGEM®-T Easy Vector kit (Promega) and clones were grouped according to their restriction fragment length polymorphism (RFLP) using the HaeIII and Tru1I restriction enzymes for actinobacteria and Tru1I and a mixture of BcnI, DpnI and PdiI restriction enzymes (MBI Fermentas) for fungi. Actinobacterial sequences were tested against chimeras using the Ribosomal Database Project II database v 8.1 CHIMERA CHECK -program (http://rdp8.cme.msu.edu/cgis/chimera.cgi?su=SSU) and fungal sequences were tested against chimeras manually in the GenBank database. Selected clones representing at least one samples from the different RFLP groups were sequenced at the Institute of Biotechnology (University of Helsinki, Finland) and compared to known sequences in the GenBank database (http://www.ncbi.nlm.nih.gov/BLAST/). The partial rDNA sequences of all clones reposted here are available under the following GenBank accession numbers: EU708341-EU708433. Each clone was assumed to represent one single operational taxonomic unit (OTU). Phylogenetic trees based on ME minimum evolution, were composed using the BioEdit -program (v. 7.0.4.1) [24] and the PAUP*-program, version 4.0, beta 10 [25]. Rarefaction analysis of the clone libraries averaged over 50 simulations were performed using the EstimatesWin800 software (version 8.0.0., http://viceroy.eeb.uconn.edu/estimates) [26].
RESULTS
Characterization of the Soil Samples
The measured soil parameters of the soil samples in the two soil profiles are presented in Table 1. The soils were generally very dry, sandy soils with low organic matter contents. An exception was the contaminated sample at the depth of 3.0-3.8.m, where the organic matter content was 40.6 % and the dry weight was only 47.6 %. This sample also had the highest PHC content, corresponding to 11 % of the organic matter content in the sample. The soil layer immediately below this fraction at 3.8-4.0 m depth contained no PHCs. This sample represented a clear separate 20 cm soil layer that differed in color from the rest of the profile.
Physical Parameters of Soil Samples in the Contaminated and Clean Soil Profiles
| Annual Mean (%)a | |||||||
|---|---|---|---|---|---|---|---|
| Sample | Depth | Dry Weight (%) | Organic Matter (%) | Petroleum Hydrocarbons (mg kgdw-1) | O2 | CO2 | CH4 |
| Contaminated | 0.0-0.5 | 94.6 | 2.2 | 500 | ndb | nd | nd |
| 0.5-1.0 | 96.2 | 0.5 | 330 | 14.6 | 7.2 | 0.0 | |
| 1.0-1.5 | 93.2 | 2.5 | 8160 | 3.5 | 13.9 | 3.4 | |
| 1.5-2.0 | 96.8 | 0.8 | 5550 | nd | nd | nd | |
| 2.0-2.5 | 95.1 | 0.9 | 12590 | 0.2 | 17.5 | 16.2 | |
| 3.0-3.8 | 47.6 | 40.6 | 44390 | nd | nd | nd | |
| 3.8-4.0 | 92.9 | 0.3 | <50c | 0.6 | 16.0 | 15.5 | |
| 4.0-4.5 | 86.0 | 0.6 | 1190 | nd | nd | nd | |
| 4.5-5.0 | 93.0 | 0.4 | 6820 | 0.5 | 16.0 | 15.4 | |
| 5.5-6.0 | 92.5 | 0.6 | 10290 | nd | nd | nd | |
| 6.1-7.0 | 82.7 | 0.4 | 140 | nd | nd | nd | |
| Clean | 0.0-1.0 | 95.3 | 0.7 | <50 | nd | nd | nd |
| 1.0-2.0 | 93.5 | 0.3 | <50 | 20.7 | 0.3 | 0.0 | |
| 2.0-3.0 | 92.5 | 0.3 | <50 | 18.9 | 2.9 | 0.0 | |
| 4.0-5.0 | 90.8 | 0.3 | <50 | 18.2 | 3.3 | 0.0 | |
| 5.0-6.0 | 79.6 | 0.9 | <50 | nd | nd | nd | |
a Annual mean in gas monitoring wells situated within 5 m distance from the sampling points [16].
b Not determined.
c Below detection limit.
Concentration of Phospholipid Fatty Acids (PLFAs) in Soil Samples
| Conc. of PLFAs in Soil [nmol (g Organic Matter)-1] | |||||
|---|---|---|---|---|---|
| Sample | Depth (m) | Total | Bacteria | Fungi* | Actinobacteria** |
| Contaminated | 0.0-0.5 | 330 | 130 | 46 | 3.9 |
| 0.5-1.0 | 620 | 320 | 11 | 14.5 | |
| 1.0-1.5 | 180 | 80 | 9 | 3.8 | |
| 1.5-2.0 | 180 | 80 | 9 | 3.1 | |
| 2.0-2.5 | 260 | 120 | 11 | 2.7 | |
| 3.0-3.8 | 10 | 5 | 1 | 0.0 | |
| 3.8-4.0 | 220 | 110 | 5 | 0.3 | |
| 4.0-4.5 | 340 | 120 | 8 | 0.6 | |
| 4.5-5.0 | 280 | 110 | 5 | 0.6 | |
| 5.5-6.0 | 290 | 90 | 6 | 0.4 | |
| 6.1-7.0 | 90 | 30 | 1 | 0.2 | |
| Clean | 0.0-1.0 | 500 | 170 | 48 | 15.5 |
| 1.0-2.0 | 240 | 100 | 46 | 3.6 | |
| 2.0-3.0 | 40 | 20 | 1 | 0.2 | |
| 4.0-5.0 | 60 | 40 | 0 | 0.4 | |
| 5.0-6.0 | 10 | 4 | 0 | 0.0 | |
* Based on the 18:2ω6 concentration.
** Based on the 10Me18:0 concentration.
Mean Values of Selected PLFAs Expressed as Mol% in Different Soil Profiles. Significant Differences Between the Groups in Tukey's B Test are Indicated by Letters
| n | Gram Positive PLFA (Mol%) | Gram Negative PLFA (Mol%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Depth (m) | i15:0 | i16:0 | i17:0 | a15:0 | 16:1ω5 | cy17:0 | 18:1ω7 | cy19:0 | ||
| Contaminated | 0-2.5 | 5 | 4 +/- 0.3 | 2 +/- 0.1 | 1 +/- 0.2 | 5 +/- 0.9a | 3 +/- 1.7 | 7 +/-1.1b | 13 +/-5.7ab | 5+/- 0.8 |
| 3.0-7.0 | 6 | 7 +/- 2.8 | 2 +/- 0.5 | 1 +/- 0.3 | 13 +/- 2.3b | 2 +/- 0.8 | 2 +/- 1.3a | 6 +/- 3.8a | 1+/- 0.8 | |
| Clean | 0-2.0 | 2 | 5 +/-0.3 | 2 +/- 0.1 | 1 +/- 0.1 | 4 +/- 0.2a | 1 +/- 0.9 | 4 +/- 0.2a | 7 +/- 1.4a | 10+/- 3.6 |
| 3.0-5.0 | 2 | 3 +/-0.8 | 2 +/- 0.5 | 1 +/- 1.2 | 3 +/- 1.4a | 8 +/- 8.2 | 8 +/- 3.4b | 31+/- 28.8b | 3+/- 0.4 | |
Closest Match of Actinobacterial Operational Taxonomic Units (OTUs) in GenBank. Percentage in Brackets Indicate the Similarity to the Closest Match
| Clean Soil | Contaminated Soil | ||
|---|---|---|---|
| % of Clone Library | Closest Match of Actinobacterial OTUs | % of Clone Library | Closest Match of Actinobacterial OTUs |
| 24 | Uncultured bacterium (88%) | 18 | Mycobacterium fallax (99%) |
| 21 | Uncultured Actinobacterium (98%) | 9 | Uncultured bacterium (99 %) |
| 14 | Uncultured bacterium (98%) | 9 | Uncultured Mycobacterium sp. (100%) |
| 8 | Uncultured Actinobacterium (91%) | 9 | Nocardia sp. (99%) |
| 8 | Uncultured bacterium (96%) | 9 | Mycobacterium sp. (99%) |
| 7 | Arthrobacter sp. (99%) | 8 | Microbacterium sp. (98%) |
| 3 | Uncultured Actinobacterium (95%) | 6 | Mycobacterium sp. (99%) |
| 3 | Uncultured bacterium (89%) | 6 | Rhodococcus sp. (99%) |
| 2 | Uncultured Actinobacterium (95%) | 5 | Microbacterium sp. (99 %) |
| 1 | Corynebacterium sp. (99%) | 3 | Tsukamurella sp. (99%) |
| 1 | Uncultured bacterium (89%) | 2 | Leucobacter sp. (99%) |
| 1 | Uncultured Actinobacterium (97%) | 1 | Uncultured bacterium (96%) |
| 1 | Uncultured Gemmatimonadetes bacterium (95%) | 1 | Microbacteriumsp. (99%) |
| 1 | Uncultured bacterium (97%) | 1 | Actinobaculum sp. (94%) |
| 1 | Uncultured bacterium (89%) | 1 | Gordonia sp. (95%) |
| 1 | Uncultured bacterium (97%) | 1 | Nakamurella multipartita (100%) |
| 1 | Uncultured bacterium (95%) | ||
| 1 | Streptomyces sp. (99%) | ||
| 1 | Leucobacter sp. (98%) | ||
| 1 | Actinobacteria (98%) | ||
| 1 | Dietzia maris (100 %) | ||
The oxygen concentration in the contaminated soil profile was reduced already at less than 1m below the soil surface and was less than 1 % in the lower soil layers where PHC concentrations were highest at 3-3.8 m. In layers with reduced oxygen levels, carbon dioxide and methane concentrations were highly elevated (Table 1). The oxygen concentration was only slightly reduced below 4 m depth at the clean sampling point and the carbon dioxide concentration was correspondingly slightly elevated. No methane was detected in clean soils.
Microbial Biomass in the Soil Profiles Based on PLFA
The total amount of PLFA was fairly constant in the contaminated soil profile down to 6 m depth (180-340 nmol g-1 org matter), except for the layer at 0.5-1.0 m depth where 620 nmol g-1 org matter was found, and the layer at 3.0-3.8 m depth in which only 10 nmol g-1 org matter was found (Table 2). The clean soil profile had a larger microbial biomass in the top layer, but the total amount of PLFAs decreased already at 2 m depth. Bacterial PLFAs were between 4 and 320 nmol g organic matter)-1, corresponding to bacterial concentrations from 3 x 108 bacterial cells (g organic matter)-1 to 2 x 1010 bacterial cells (g organic matter)-1 when using the conversion factor 1.4x10-17 mol PLFA bacterial cell-1 [18]. The bacterial biomass at depths below 2.0 m was about 5 times higher in the contaminated profile compared to the clean profile, except for the layer 3.0-3.8 m in the contaminated profile where the bacterial biomass was very low. The reason for this low value is unclear; it could be a real value, or an artifact as a result of inefficient PLFA extraction due to presence of high amounts of PHCs in this sample (40.6 % organic matter compared to the surrounding layers that had 0.9 and 0.3 % organic matter, see Table 1). The fungal biomass in the top layer was similar in the two profiles, 47 nmol g-1 organic matter, but it decreased more rapidly with depth in the clean profile compared to the contaminated profile. At 3-6 m fungal PLFAs were frequently detected in the contaminated soil profile. Similar as for the fungi, the actinobacterial indicator 10Me18:0 was highest in the upper 2 m of both soil profiles and was only found in low amounts below 2.5 m (Table 2).
Principal component analysis (PCA) based on mol% of the PLFAs separated the samples into three distinct groups along the principal component 1 (PC1), which explained 22 % of the variation in the samples (Fig. 1). The contaminated soil samples from the lower levels (≤3 m) clustered to the left in the PC plot, while samples from the upper contaminated soil samples and from the clean soil were found to the right.
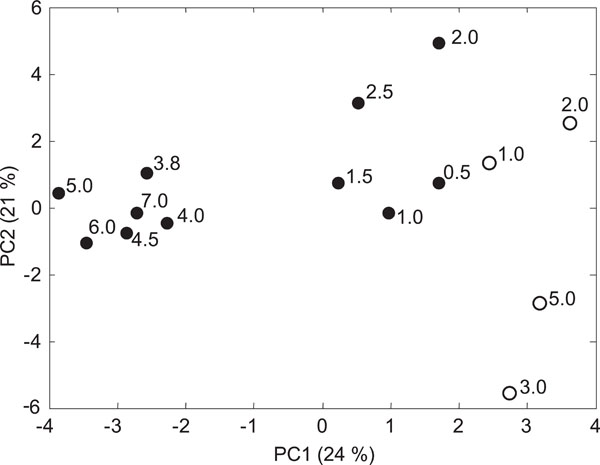
Biplot of soil samples in principal component analysis (PCA) based of the molar ratios (mol%) of PLFAs from the contaminated soil profile (filled symbols) and the clean soil profile (open symbols). Numbers next to the symbols indicate the higher limit of the corresponding soil profile. ω
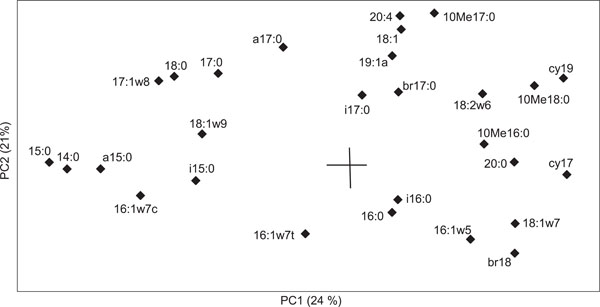
Biplot of loading values for different PLFAs in principal component analysis.
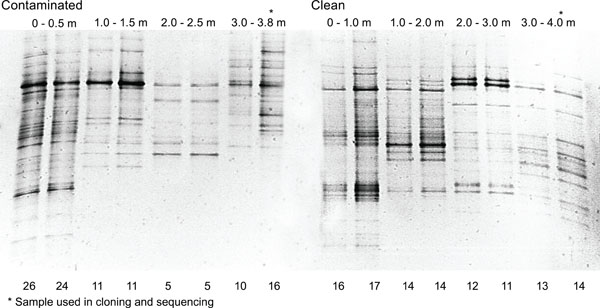
DGGE profiles of Actinobacteria in contaminated and clean soil profiles. The result from two parallel DNA extractions are shown. Number of bands detected in the soil profiles are indicated below the figure at each band.
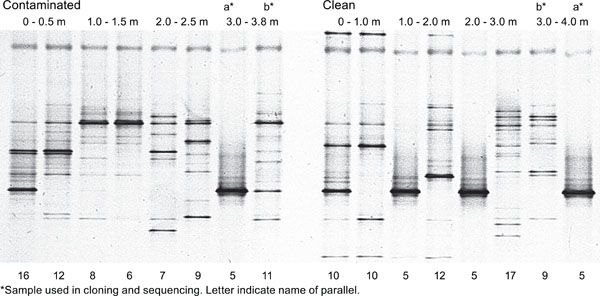
DGGE profiles of fungi in contaminated and clean soil profiles. The result from two parallel DNA extractions are shown. Numberof bands detected in the soil profiles are indicated below the figure at each band.
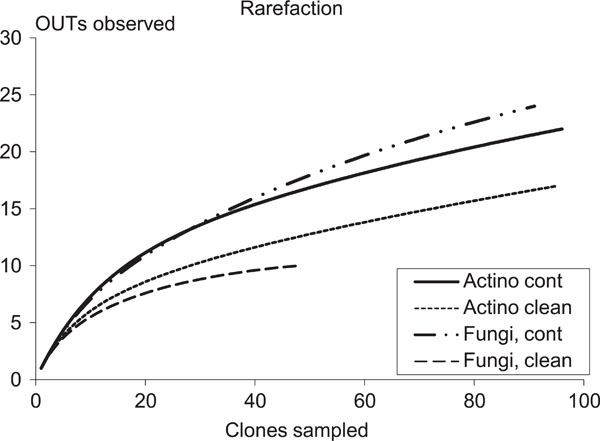
Accumulation curves clone libraries. Curves are averaged over 50 simulations using the computer program EstimateS.
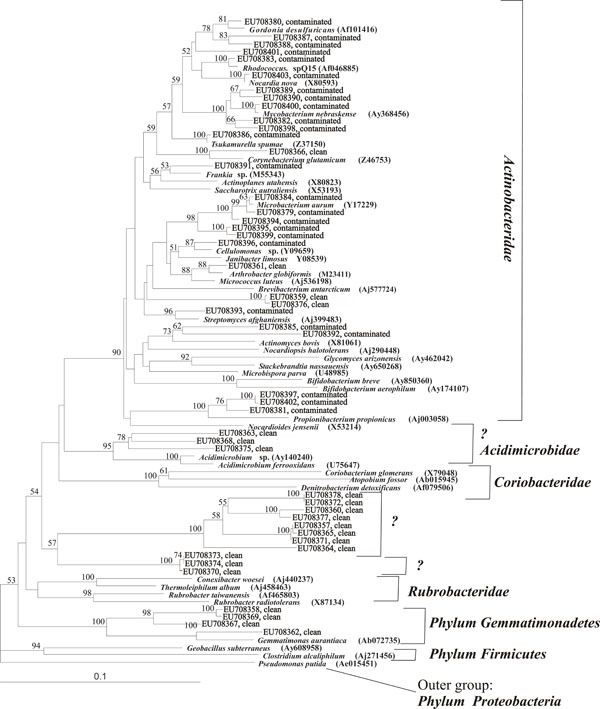
Phylogenetic tree of Actinobacterial OTUs as isolated from contaminated and clean soil samples from a depth of 4 m in relation to selected OTUs from Genbank (accession numbers of GenBank OTUs are indicated in brackets). OTUs are reported as Genbank accession numbers. Bootstrap values (%) are indicated for the branches which were similar in more than 50 % of the 1000 regenerated trees.
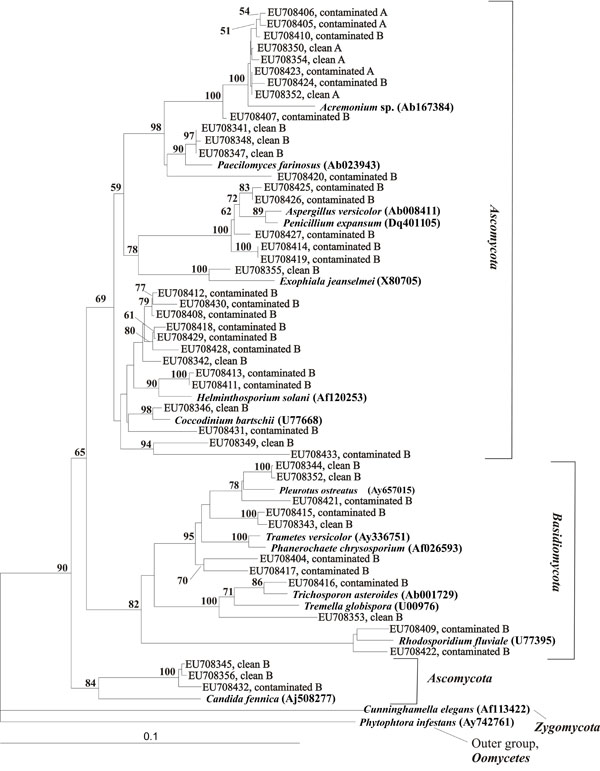
Phylogenetic tree of fungal OTUs isolated from contaminated and clean soil samples from a depth of 4 m in relation to selected OTUs from Genbank (accession numbers of GenBank OTUs are indicated in brackets). OTUs are reported as Genbank accession numbers. Bootstrap values (%) are indicated for the branches which were similar in more than 50 % of the 1000 regenerated trees.
The most important grouping variables for principal component 1 (PC1) were the PLFAs cy17:0, cy19:0 and 18:1ω7, indicative of Gram negative bacteria; 10Me18:0 indicative of actinomycetes, and 18:2ω6 indicative of fungi, which were all found at higher proportions in the clean soil and in the upper part of the contaminated soil whereas the PLFAs 15:0, 14:0, a15:0, and 18:0 were found at higher proportions in the lower layers of the contaminated soil (Fig. 2). Samples from the two lowest sampling layers (2.0-3.0 m and 5.0-6.0 m) from the clean soil grouped separately from the rest of the samples according to principal component 2 (PC2). The mol% values of two of the four indicator PLFAs for Gram positive bacteria were higher in the deeper layers compared to the upper layers of the contaminated soil profile (Table 3). The mol% values of all four indicator PLFAs for Gram negative bacteria were lower in the deeper contaminated soil layers than in the upper soil layers of the contaminated soil profile. In the clean soil profile on the other hand, the mol% of indicator PLFAs for Gram negative bacteria increased with soil depth in three cases out of four. Although statistical differences between the upper (0-2.5 m) and lower (3.0-7.0 m) are scarce, this indicates that gram positive bacteria may be more dominant in the lower soil layers of contaminated sites but probably not in clean subsurface soils.
Microbial Community Composition Based on Extracted DNA
DNA was extracted from the four uppermost soil layers of the two soil profiles. DNA levels were low in all samples and decreased with depth, corresponding to the decrease in microbial numbers indicated by PLFAs (data not shown). The highest values were found in the most contaminated soil layer at the depth of 3.0-3.8 m and the lowest yield in the deepest soil sample of the clean soil.
The actinobacterial DGGE profiles were different in the contaminated and the clean soil profiles. In both soil profiles, the uppermost soil contained most bands; approximately 25 bands were detected in the contaminated soil sample and approximately 16 bands were detected in the clean soil sample (Fig. 3). The number of bands decreased with soil depth especially in the contaminated soil profile. On the other hand, in the deepest soil sample (3.0-3.8 m), containing much organic matter and highest PHCs, but reduced oxygen, the number of bands was slightly higher than in the previous layer (2.0-2.5 m). In the clean soil profile, where the oxygen level did not decrease with depth, the decrease in number of bands was less pronounced (Fig. 3). Actinobacterial DGGE profiles from parallel DNA extractions were similar indicating that DNA extraction and PCR procedures were reproducible.
The number of bands in fungal DGGE profiles did not significantly decrease with soil depth (Fig. 4). To our surprise, the parallels of the fungal DGGE profiles differed and often one of the parallel samples contained only one dominating band. This was the case for three of the four soil layers studied in the clean soil. In the contaminated soil layer, one strong band appeared in one of the parallels at 3.0-3.8 m depth but not in the other three soil depths studied.
Species Composition of Soil Layers from a Depth of 4 m
Clone libraries were constructed from samples taken at 3-4 m depth. A total of 96 and 95 actinobacterial clones were selected for further studies from the contaminated and clean soil layers, respectively. From the two replicate fungal clone libraries a total of 15 and 91 clones from the contaminated soil and 14 and 48 from the clean soil were selected. The clone libraries represented by only 14 or 15 clones studied were derived from the fungal replicate sample A with only a few bands in the DGGE profile. The leveling off of the rarefaction curves (plots of cumulative number of OTUs as a function of a clone number) indicate that the majority of the OTUs in the sample were represented in the clone libraries (Fig. 5).
A total of 16 and 21 different actinobacterial OTUs were identified by RFLP and sequenced in the clean soil and contaminated soil samples, respectively. The major part (90%) of the actinobacterial OTUs from the clean soil layer at 4 m matched only to uncultured bacteria or uncultured actinobacteria and the match was often less than 97% (Table 4). The three most frequently found OTUs, of which all were related to uncultured bacteria, represented 60 % of the clone library. Some of the OTUs from clean soil formed two phylogenetically distinguished groups separate from any reference OTUs in the phylogenetic tree between the subclasses Coriobacteridae and Rubrobacteridae (Fig. 6). It is possible that these groups of actinobacteria may represent new subclasses. Other uncultured OTUs grouped phylogenetically with the subclasses Acidimicrobidae, Actinobacteridae and one OTU with Gemmatimonadetes, which is not considered an actinobacterial group. The three clones related to Acidimicrobium may also represent a separate group of bacteria. The OTUs from the contaminated soil all grouped with the subclass Actinobacteridae (Fig. 6). Contrary to the OTUs in clean subsurface soil, most OTUs at the contaminated soil sample from the same depth matched cultured actinobacterial phyla. Six types of OTUs, of which only two matched uncultured strains, represented 60 % of the clone library and 30 % of the clone library of the contaminated sample matched with Mycobacterium (Table 4).
A total of 19 and 10 different fungal OTUs were identified by RFLP in the contaminated and the clean soil samples, respectively. From both fungal clone libraries B, which were expected to have higher diversity based on DGGE results, OTUs grouped phylogenetically with the Ascomycota and Basidiomycota (Fig. 7). The RFLP groups detected in the clean soil sample B were fewer than in the contaminated soil. As expected, all OTUs in the contaminated soil and clean soil samples A represented the same OTU and they matched to Acremonium sp and Paecilomyces sp in GenBank. Phylogenetically these clones were closely related (Fig. 7). In the more diverse B sample, OTUs relating to Acremonium sp and Paecilomyces sp was only found in the contaminated soil, where it represented 10 % of the clone library.
DISCUSSION
Two different soil profiles down to the depth of 7 m were examined in this study. The profiles originated from a 30-year-old site contaminated with PHCs and from a clean site next to the contaminated site. The concentration of oxygen in the contaminated soil profile was strongly reduced in soil layers below 1 m depth but not in the clean soil profile. The microbial biomasses based on bacterial and fungal PLFAs varied from 90-330 nmol (g organic matter)-1 in the uppermost 2 m of both soil profiles to less than 10 nmol (g organic matter)-1 in the lowest part of the clean soil profile. This is ten times less than have earlier been reported for top soils in forest soil [27] but similar to subsoils (3-4 m) contaminated with benzotriazole [3]. At soil depths below 2 m the microbial PLFA concentrations in the contaminated soil was more than 5 times higher than in the corresponding clean soil layers at the same depth, despite the fact that the availability of oxygen was very low in the contaminated soil profile. Bacterial numbers in the deepest soil layers estimated based on the PLFA data [18] were 1.2 x 107 bacterial cells (g dry weight)-1 or 3.1 x 109 bacterial cells (g organic matter)-1 in the contaminated soil layer at 7 m and 3.3 x 106 bacterial cells (g dry weight)-1 or 3.9 x 108 bacterial cells (g organic matter)-1 in the clean subsurface soil at 6 m. Information on bacterial numbers in deep subsurface is scarce but recent reports indicate that bacterial numbers of specific groups in deep subsurface soils may be considerable [2]. The impact of PHC as potential carbon source explains the larger bacterial numbers in the anaerobic contaminated soil compared to the clean soil [9].
Principal component analysis based on PLFA data (Fig. 1) demonstrated that the microbial community composition in the clean soil profile differed from that of the contaminated soil profile and also that the microbial community in the upper 2.5 m layers of the contaminated soil profile differed from the lower soil layers. This grouping cannot only be due to the redox situation in the soil as oxygen depletion was recorded already below 1 m. Our results indicate that Gram positive bacteria increased and Gram negative bacteria decreased with depth in the contaminated soil profile. Similar observations have earlier been reported several times for the upper 2-3 m of soil profiles [9, 28, 29]. In both soil profiles, the amount of fungi decreased with depth but in contaminated soil fungal PLFAs were detected also at 3-6 m depth. These results indicate that Gram positive bacteria may be relevant for bioremediation purposes in subsurface soils. In the clean soil profile, the Gram negative bacteria did not decrease with soil depth but instead increased in samples below 3 m. The reason for this remains unclear. The proportion of actinobacteria found of in the present study (0-1 % of the total bacteria, based on PLFA values) was in accordance with other investigations showing similar proportions of actinobacterial clones in clone libraries from anoxic contaminated subsurface [15, 30].
The composition of actinobacterial communities based on rDNA sequences in DGGE analyses were surprisingly diverse in both subsoil profiles. It is possible that the limited oxygen conditions in the contaminated profile caused the observed decrease in DGGE bands at 1-3.8 m. Many OTUs in the contaminated soil sample matched to Mycobacterium. Mycobacterium strains may metabolize polyaromatic hydrocarbons (PAHs) at low nutrient conditions [31] and their specific genes involved in n-alkane degradation have been detected in contaminated Alpine soils [10]. In the clean soil profile the decrease in actinobacterial OTUs was less pronounced. The actinobacterial community was different in the clean and the contaminated soil profile. It is especially noteworthy that most of the actinobacterial OTUs detected in the clean soil at 3-4 m represent bacteria that have not been cultured and that some of these seem to represent new, previously undescribed subclasses of Actinobacteria. Three of the isolated OTUs showed similarity to the subclass Acidimicrobidae, which also is a relatively unknown group, because of the small amount of isolates in this subclass [32]. It thus appears that there was an uncharacterized community of actinobacteria in the subsurface which may be unexploited for different applications.
We found several different fungal rDNA sequences in DGGE analyses also at lower depths, both in clean soil and contaminated soil with reduced oxygen concentrations. The closest matches for most of these in the GenBank database were only obtained to the family level or higher. More specific taxonomic grouping was not achieved for most of the OTUs because they matched with several different fungal groups with same similarity percentages. The 18S rDNA primers used in this study may not be specific enough to give a better distinction and inclusion of primers for ITS sequences could have given more specific identification. It is interesting that often one dominating group, related to the Acremonium sp. and Paecilomyces sp. was found both in contaminated and in clean soil samples. A fungal isolate belonging to Acremonium sp. was recently found in Malaysian crude oil [33] and was earlier shown to degrade hydrocarbons in batch cultures [34]. Contrary to the analyses of actinobacterial DNA, the fungal replicate samples in the DGGE profiles did not show similar band patterns. The distribution of fungal species in soil is known to be spatially clustered leading to hot spot distributions due to their hyphal growth [35]. It has been observed that small soil samples exhibited a larger variation in fungal community composition between replicas than larger soil samples [36]. It was concluded, that samples <1 g are convenient for obtaining reproducible bacterial fingerprinting, but samples of at least 1 g are required for obtaining reproducible fungal fingerprinting.
The use of molecular tools for characterization of the microbial community in subsoils enabled detection of groups of micro-organisms that have been considered to be strictly related to aerobic environments. Similar observations have been reported for aerobic fungi in anaerobic sediments [37]. The use of specific primers for the actinobacteria group allowed a better insight into which actinobacteria are present in this environment, because better resolution is obtained than when using general bacterial primers as in most other analyses of microbial community composition. The fact that many clones affiliated with mycobacteria were found in the contaminated subsurface could suggest that these organisms may possess an unknown ability to degrade contaminants under aerobic or microaerophilic conditions. The methods used here do not distinguish between active microbial cells and cells that are in a resting stage. Both of the groups studied here, the Actinobacteria and the fungi, often form resting stages like spores to endure unfavorable environmental conditions. Single spore cells are however unlikely to be detected with these methods, because several copies of the target DNA is usually needed for detection with these methods in environmental samples. It should also be kept in mind that the microbial DNA profiles in the upper soil layers often differed from the microbial DNA profiles in lower soil levels. It is likely that some organisms establish more easily than others in the lower layers, thus also leading to differences in community composition. Therefore the upper soil layer is not a likely a source for the micro-organisms in the lower part of the soil profile.In this work we show that the cell number decreases with soil depth but also that microbial community size and species number (determined as OTUs) in soil layers below 2 m are considerable both in clean and PHC contaminated soil profiles. It is well known that the amount of molecular markers does not reflect the total diversity of populations [38]. The microbial communities differed between the two very different subsurface environments and they differed also with depth. Low oxygen concentration and high PHC concentration did not restrain the diversity of these groups of micro-organisms at this 30-year-old contaminated site. In the clean soil profile, most of the actinobacterial OTUs matched to uncultured strains. Some of these OTUs might even represent a new subclass in the actinobacterial phyla reflecting that little is known about the whole microbial diversity in the biosphere. The metabolic state of the micro-organisms described in this work was not studied. More effort is needed to gain insight into the in situ activities of microbial communities in subsurface soils. The results indicate that fungi and actinobacteria may be important components of the microbial communities in hydrocarbon contaminated and oxygen limited subsoils and more knowledge about these groups may help to develop more effective inocula to be used in soil bioremediation. New information from genomics may also help to reveal if actinobacteria and fungi possess hitherto unknown abilities to degrade contaminants.
ACKNOWLEDGEMENTS
We thank Timo Vänni and Helena Tuomola for skilful assistance with the figures. This work was funded by the EU/LIFE programme (number LIFE 03 ENV/FIN/000250) and by the Microbiology and bioremediation in cold climate (MECBIO)-NordForsk funded network.

