All published articles of this journal are available on ScienceDirect.

Proteome Analysis of Leptospira interrogans Virulent Strain
Authors Info & Affiliations
Abstract
Leptospirosis is a worldwide zoonotic infection of human and veterinary concern. Caused by pathogenic spirochetes of the genus Leptospira, the disease presents greater incidence in tropical and subtropical regions. The identification of proteins that could be involved in the bacteria host interactions may facilitate the search for immune protective antigens. We report the proteomic analysis of Leptospira interrogans serovar Pomona virulent strain LPF cultured from kidney and liver of infected hamsters. Total protein extracts were separated by two-dimensional gel electrophoresis (2-DE), 895 spots were analyzed by MALDI-TOF mass spectrometry (MS), and 286 were identified as leptospiral proteins, corresponding to 108 distinct proteins. These proteins are allocated in all the bacterial cell compartments and are distributed in every functional category. Furthermore, the previously described, known outer membrane proteins, OmpL1, LipL21, LipL31, LipL32/Hap-1, LipL41, LipL45, LipL46, LruA/LipL71, and OmpA-like protein Loa22 were all recognized. Most importantly, this research work identified 27 novel leptospiral proteins annotated as hypothetical open reading frames (ORFs). We report for the first time an array of proteins of the Leptospira expressed by virulent, low-passage strain. We believe that our studies, together with the genome data will enlighten our understanding of the disease.
INTRODUCTION
The spirochete Leptospira interrogans is a highly invasive pathogen and the causal agent of leptospirosis, one of the most widespread zoonosis of human and veterinary concern [1-5]. The leptospires chronically infect domestic and wild mammalian species that harbor the bacteria in their renal tubules, shedding them through the urine into the environment, contaminating soil, water, and re-infecting other animals. Humans are accidental hosts in the transmission process of leptospirosis [5, 6]. The leptospires enter the body via abrasions on the skin, or enter actively through mucosa, spreading to any tissue, but specifically colonizing the kidneys and liver. Fever, headache, anorexia, muscle pain, and constipation are the general symptoms of the disease. About 5-15% of the cases evolve more severely, presenting hemorrhages with renal and hepatic failure - a condition known as Weil’s syndrome, with a mortality rate of 5-40% [4, 5]. Leptospirosis also has a great economic impact, since the disease affects the livestock inducing abortions, stillbirths, infertility, reduced milk production, and death [5]. Despite its importance, the aspects of pathogenesis, virulence, and invasion processes by which the leptospires infect the hosts and initiate tissue colonization are poorly characterized.
The genomic sequences of five strains of Leptospira, four pathogenic [7-9] and one non-pathogenic [10], are now available. The genomic comparison and analysis in the pathogenic species revealed many hypothetical open reading frames (ORFs) and proteins with unknown functions that might be involved in the pathogenesis and virulence of the leptospires. Although the genomic tools are important to reveal the general aspects of the organism, it does not characterize which proteins are being expressed under a certain condition. For instance, it has already been demonstrated that some leptospiral virulence factors are down-regulated after several passages in the culture medium [11-13]. Accordingly, proteomics is being used in an attempt to solve some of these questions; however, so far, only a few Leptospira proteomic or immunoproteomic studies have been published [14-18]. Furthermore, these works have focused on the identification of Triton X-114-derived membrane proteins [16, 19], affinity capture of biotinylated membrane proteins [14], outer membrane vesicles [15], or in the case of immunoproteomics, the identification of proteins that are reactive with immune mouse serum [17] or serum from chronically infected rats [20].
In this study, we report the analysis of the protein expression profile of L. interrogans serovar Pomona strain LPF cultured, low-passage, from kidney and liver of infected hamsters. The choice of the serovar was due to the well established virulence of this strain in our laboratory. We identified by 2-DE/mass spectrometry (MS) 286 leptospiral proteins, corresponding to 108 distinct proteins. We confirmed the expression of 27 novel proteins that are genome annotated as hypothetical. Additionally, we identified some uncharacterized predicted outer membrane proteins, which are a class of proteins that have been the focus of the leptospirosis research worldwide.
MATERIALS AND METHODS
Bacteria
The virulence of the L. interrogans serovar Pomona strain LPF was maintained by iterative passages in Golden Syrian hamsters. Recently weaned hamsters were intraperitonially infected with 500 µL of approximately 1 × 104 virulent leptospires. The animals were sacrificed after the appearance of symptoms, such as loss of weight and mobility (approximately 5 days post-infection). Kidney and liver were removed and macerated. The leptospires recovered from these organs were cultured at 28°C in semi-solid EMJH-modified medium supplemented with 10% rabbit sera, followed by sub-culture in liquid EMJH (Difco®- USA) supplemented with 10% rabbit sera, until the density of approximately 108 cells/mL was reached.
Whole-Cell Protein Extracts
The culture was harvested by centrifugation at 12800× g at 4°C for 10 min. Pellet was washed (×5) by resuspension in 35 mL phosphate-buffered saline (PBS) containing 5 mM MgCl2, followed by centrifugation under similar conditions described earlier. The pellet was weighed and resuspended in DeStreak Rehydration Solution (GE Healthcare, USA) at the proportion of 5 µL/mg of the bacteria. The cells were lysed by vigorous vortexing followed by mechanical pressure through a syringe. The cellular debris was separated by centrifugation at 20800× g for 10 min at room temperature and the supernatant was collected. Total protein content was determined according to the Bradford method (Pierce Biotechnology, USA), following the manufacture’s protocol. Bovine serum albumin (BSA) was used to generate a standard reference curve (0-10 µg). The protein solution was mixed at the appropriate proportions with the Bradford reagent dye, and after 30 min of incubation at room temperature, the readings were taken at 595 nm. The protein concentration from each sample was calculated based on BSA standard absorbance curve. Samples of 700 µg of protein were adjusted to 340 µL with DeStreak Rehydration Solution (GE Healthcare, USA), along with 0.8% (v/v) IPG buffer, with a pH range of 3-10 (GE Healthcare). The protein extracts were obtained in triplicate from bacteria cultured from kidney and liver of infected animals.
Two-Dimensional Gel Electrophoresis (2-DE Gels)
First-dimension isoelectric focusing was performed using the IPGphor-System (GE Healthcare, USA), and the second dimension was conducted on the Ettan DALTsix system (GE Healthcare). The IPG gel strips (18 cm) with a linear separation of immobilized pH ranging from 3 to 10 were rehydrated directly with the solubilized samples. The strips were covered with mineral oil to prevent dehydration and oxidation, and the process was carried out overnight at room temperature in the Immobiline Dry-Strip Reswelling Tray (GE Healthcare). The focusing protocol was 30 V for 180 Vh, 150 V for 300 Vh, 350 V for 350 Vh, 500 V for 500 Vh, 1000 V for 1000 Vh, 3000 V for 3000 Vh, and 5000 V for 65000 Vh, with a 50 µA/strip maximum-setting at 20°C. The strips were equilibrated twice (reduced and alkylated) for 15 min in 15 mL equilibration solution (0.05 M Tris-HCl, pH 8.8, 6.0 M urea, 30% [v/v] glycerol, and 2% [w/v] SDS), first with the addition of 1% DTT, and finally with 2.5% iodoacetamide. After equilibration, the strips and the molecular-mass marker proteins were attached to the 12% SDS-PAGE, 1 mm thickness, using 1% agarose. The electrophoretic conditions were as follows: 5 W/gel for 30 min and 17 W/gel until the end of the running. The gels were stained for 24-48 h using Coomassie Blue R350 (PhastGel Blue R - GE Healthcare) under gentle agitation, and then were rinsed with ultrapure water (MilliQ grade). The destaining of the spots was carried out in the solution of 40% (v/v) ethanol and 10% (v/v) acetic acid. The gels were digitalized in transparency mode, and analyzed with Image Master-2D Platinum version 6.0 software (GE Healthcare, USA). The 2-DE gels were performed in triplicates for the protein extracts of leptospires cultured from kidney and liver of infected hamsters.
Mass Spectrometry
The samples were analyzed by MALDI-TOF (Matrix Assisted Laser Desorption Ionization-Time of Flight) MS, using α-cyano-4-hydroxycinnamic acid as the matrix on an Ettan MALDI-TOF/Pro instrument (Amersham Biosciences, USA). Briefly, the gel spots were isolated and dehydrated with acetonitrile (ACN). Then, the treated spots were rehydrated with 40 ng/μL trypsin solution in 50 mM ammonium acetate, and the reaction was carried out overnight at 37°C. The tryptic peptides were extracted from the gel spots using an H2O/ACN/TFA (1:1:0.05) solution and this procedure was repeated 3X. The peptide solutions were concentrated and desalted with ZipTip C-18 pipette microcolumns (Millipore) prior to analysis. The samples were pre-mixed with the matrix solution (1:1 [v/v]) and approximately 0.5 μL was dropped on the sample loader and let to dry over the bench. The MS was performed on the reflectron mode and internal calibration (trypsin autolysis peptide). External calibration was also carried out in case the trypsin peptides were not present or clear.
Database Searches
The MALDI-TOF obtained mass lists were analyzed by peptide mass fingerprinting (PMF) algorithms on the server MASCOT (http://www.matrixscience.com/cgi/search_form.pl?FORMVER=2&SEARCH=PMF) for matches with known protein sequences deposited on the public or proprietary databases. The searches were performed against MSDB non redundant and L. interrogans serovar Copenhageni databases without any MW or pI restrictions. One missed cleavage site per peptide was allowed, and an error tolerance of ±0.2 Da on the mass measurement was used for the search and modifications such as, carbamidomethylation of cysteines and random oxidation of methionines were taken into account. The positively identified protein matches were considered to be those that were statistically significant by the probability-based MOUSE scoring of the server MASCOT (score>42), which takes into account a value of p<0.05 for the random matches.
Additional Bioinformatic Analyses
The indentified L. interrogans proteins were further analyzed by the bioinformatics tools. The web-based tool P-CLASSIFIER [21] that considers five primary localization sites of Gram-negative bacteria (cytoplasm, extracellular space, inner membrane, outer membrane, and periplasm) was used to predict the subcellular localization of the identified proteins (http://protein.bii.a-star.edu.sg/localization/gram-negative/), taking into account the greatest probability. The LipoP server [22] (http://www.cbs.dtu.dk/services/LipoP/) was used to predict the lipoproteins and discriminate between lipoprotein signal peptides (SpII), other signal peptides (SpI), and n-terminal membrane helices (TMH) that might be present in the proteins. This program is specific for Gram-negative bacteria and is based on the lipobox consensus sequence. For the purpose of analysis of functional category of the identified proteins, the genomic database of L. interrogans serovar Copenhageni (http://aeg.lbi.ic.unicamp.br/world/lic/), (http://cmr.jcvi.org/cgi-bin/CMR/GenomePage.cgi?org=ntli03) and the Entrez Gene of National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/sites/entrez?db=gene) were used.
All animal studies were approved by the Ethics Committee of the Faculdade de Medicina Veterinaria, Universidade de São Paulo, Sao Paulo, Brazil.
RESULTS
Two-Dimensional Gel Electrophoresis (2-DE Gels)
Protein extracts were fractioned by 2-DE gels, focalized in the pH range of 3-10 and molecular mass separated in 12% acrylamide gels. Representative Coomassie Blue R stained gels, from three independent experiments, of protein extracts of Leptospira cultured from kidney and liver are shown in Fig. (1), where some identified proteins are illustrated. The protein spots were observed to be mainly distributed between de pIs 5-7 and were almost absent in the basic or acid extreme regions of the strips (Fig. 1). Our data are in agreement with the published studies that have shown leptospiral-protein focalization between pIs 4 and 7 [19], including proteins of whole Leptospira [2].
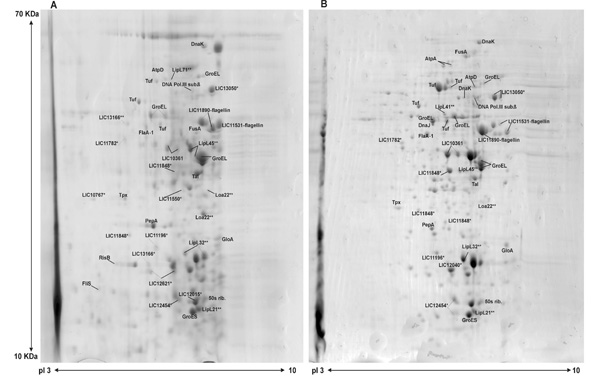
Representative 2-DE gels of L. interrogans serovar Pomona protein samples. Whole-cell protein extracts of virulent Leptospira cultured from kidney (A) and liver (B) were separated by 2-DE, with pH 3-10 and molecular mass of 10-70 kDa. Protein spots were excised for identification by MALDI-TOF MS. Examples of known leptospiral proteins, including some characterized lipoproteins (**) and some novel hypothetical proteins (*) identified in this work are depicted.
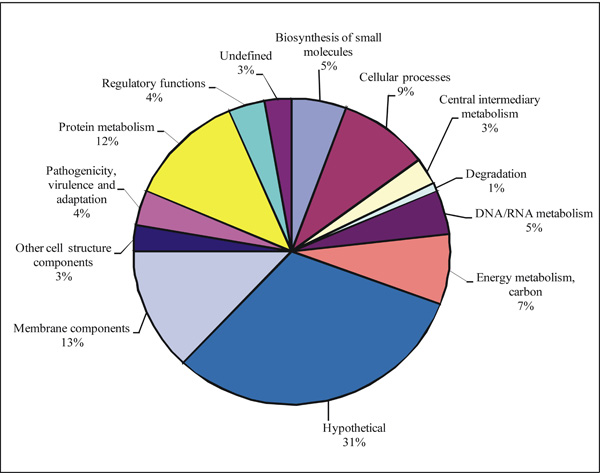
Functional classification of the proteins of L. interrogans serovar Pomona. Distribution of proteins identified by 2-DE/MALDI-TOF MS of virulent leptospires, cultured from kidney and liver of infected animals. Categorization was based on the genome annotation of L. interrogans serovar Copenhageni genome (http://aeg.lbi.ic.unicamp.br/world/lic/).
Mass Spectrometry
All the isolated and visible major protein spots were excised from each gel and were separately processed for MS. After MALDI-TOF MS, the obtained PMF mass lists were searched and compared against the L. interrogans serovar Copenhageni genomic database for the identification of the respective leptospiral proteins, and the NCBI for the general protein match. From the 6 2-DE gels obtained, a total 895 spots were analyzed, resulting in positive identification of 286 spots (31.9 %) as leptospiral proteins. The 108 recognized proteins are representative of all functional categories used in genome annotation, and their respective percentages are depicted in Fig. (2). The proteins were assigned in: (a) biosynthesis of small molecules, (b) cellular processes, (c) central intermediary metabolism, (d) degradation, (e) DNA/RNA metabolism, (f) energy metabolism and carbon, (g) hypothetical, (h) membrane components, (i) other cell structure components, (j) pathogenicity, virulence, and adaptation, (k) protein metabolism, (l) regulatory functions, and (m) undefined category. The distribution in the functional categories of the 108 proteins identified in L. interrogans showed that the allocation of the proteins is similar to all categories, with the highest number of proteins being presented in the hypothetical category (Fig. 2). This data is very important, because it validates the expression of these leptospiral proteins with unknown function. A complete list with general analysis of identified proteins of L. interrogans serovar Pomona cultured from kidney and liver of infected animals is presented in Table 1 (supporting information). Sequences of matched peptides of each identified protein are shown in Table 2, supporting information.
Bioinformatics Analysis
The identification of the surface proteins is an important theme of the current leptospiral research. Owing to their location, the leptospiral surface proteins are expected to be relevant in the host-cells interactions in the context of pathogenesis and their potential ability to promote heterologous immunity. Predicted membrane proteins, according to P-CLASSIFIER program, with or without signal peptidase I or II cleavage sites, as predicted by LipoP program, are compiled in Table 1 (supporting information). The analysis of the subcellular localization of the proteins, as predicted by P-CLASSIFIER [21] revealed that the identified proteins are allocated in all the bacterial cell compartments: cytoplasm (78), periplasm (8), inner membrane (4), outer membrane (14), and extracellular (3) (Table 1, supporting information).
The 108 identified proteins in L. interrogans serovar Pomona were also observed in L. interrogans serovars Lai and Copenhageni genomes [7, 8, 23], 99 of them were conserved in both the strains of L. borgpetersenii serovar Hardjo and 80 were present in L. biflexa serovar Patoc sequenced strains [9, 10]. Protein conservation among these later strains is depicted in Table 3 (supporting information) that shows (in gray) that 8 of the hypothetical proteins validated in this work are absent in the saprophytic non-pathogenic L. biflexa.
DISCUSSION
It is well known that leptospires lose their ability to infect after a long period of cultivation. This may be owing to the differential expression of essential virulence factors that could be suppressed according to the environmental conditions [5]. Recently, proteins that are down-regulated during the culture-attenuation process and up-regulated in conditions that approximate the host environment have been identified [12, 13]. Our work aimed to evaluate the global picture of proteome expression of virulent, low passage L. interrogans serovar Pomona sub-cultured from the kidney and liver of infected animals. Under these conditions we succeeded to have high cellular density and therefore sufficient amount of proteins to perform the proteomic analysis. In addition, it is well documented that one subculture preserves the virulence of Leptospira [5, 12, 13, 24].
A total 895 spots were analyzed, resulting in positive identification of 286 spots (31.9 %) as leptospiral proteins. The 286 spots identified by mass spectrometry corresponded to 108 different leptospiral proteins, some of them being present in multiple spots with pI and/ or molecular-mass variations. The multiplicity of the spots might be due to protein isoforms resulting from post-translational modifications or protein degradation during the 2-DE procedure, and have also been observed in other proteomic studies [14, 16, 19, 25]. The total number of proteins detected in this study by 2-DE gels coupled with MALDI-TOF MS is not high when compared with the total proteins predicted in the pathogenic strains by genome sequencing [7-9, 23]. This is probably due to the limitation of 2-DE such as, protein solubilization in aqueous media, the pH range for isoelectric focusing, the amount of protein expressed and the dynamic range of 2-DE electrophoretic separation allied to the limitation of MS method that does not always identify the proteins regardless of the noticeable amounts exhibited by protein staining. This is possibly a consequence of site-specific trypsin used in spot gel-derived for proteinase digestion [15]. Additional identification of leptospiral proteins may be improved by the use of other pH gradients for isoelectric focusing and by alternative proteinase cleavage conditions by the use of MS/MS compatible instruments.
The previously described leptospiral membrane proteins identified using Triton X-114-extract OMPs [19], biotinylated-surface proteins [14], outer membrane vesicles [15], and in outer membrane proteome of L. interrogans serovar Copenhageni [16] were recognized in this study: OmpL1 (LIC10973) [26], LipL31 (LIC11456) [2], LipL32/Hap-1 (LIC11352) [27, 28], LipL41 (LIC12966) [29], LipL45 (LIC11643) [30], LipL21 (LIC10011) [31], LruA/LipL71 (LIC11003) [32], OmpA-like protein Loa22 (LIC10191) [33]. In addition to these proteins, our work also identified LipL46, a recently characterized lipoprotein (LIC11885) [34] and, most importantly, 5 novel hypothetical SpI- and SpII-containing proteins (Table 1). However, the leptospiral immunoglobulin-like A (LigA) protein, detected in TX-114-extracted proteins [16] was not observed in our study. Additionally, the 24-kDa LfhA/ Lsa24/LenA [35-37] and Lsa21 proteins [13] were not identified. Separation of proteins by 2-DE is limited for large and highly hydrophobic proteins that are difficult to solubilize, and that might be the case of the high molecular-mass LigA protein [11]. In spite of intense studies, majority of leptospiral membrane proteins and lipoproteins identified in the genome sequences [7-9, 23] were still assigned as hypothetical proteins of unknown function. The proteome expression of these proteins identified in virulent, low-passage Leptospira validates their existence within the bacteria, and allows some speculation of their possible role in the process of leptospiral pathogenesis. We have identified 34 hypothetical-annotated proteins, being 7 of them previously validated by Nally et al. [15] and Sakolvaree et al. [17] (see Table 1, supporting information). The 27 novel proteins comprise 2 SpI-containing proteins, 3 predicted SpII lipoproteins and 2 proteins containing TMH domains, as predicted by the Lipo-P program [22]. Additionally, we have identified 7 proteins related to the flagella structure and the previously characterized chaperones GroES/Hsp10, DnaK (Hsp70), GroEL, Hsp15 [14, 38-40]. As mentioned earlier, some proteins appeared in more than one spot, such as the known chaperones, DnaK (LIC10524) and GroEL (LIC11335) that were identified in 2 and 5 spots, respectively. The elongation factor Tu (LIC12875) was identified 10 times in one gel and 5 times in the other. Furthermore, the membrane lipoproteins OmpA-like (LIC10191), LipL32 (LIC11352), LipL45 (LIC11643), and LipL41 (LIC12966), the flagellins LIC11531 and LIC11890, the ATP synthase subunits A and B (LIC11241 and LIC11243), and the hypothetical proteins LIC11848, LIC12621, LIC13050 and LIC13166 were also the proteins observed in the multiple spots over the gels. In the case of LipL32, distinct molecular mass forms and pI isoforms were shown to comprise approximately 31% of the gel-separated protein spots of Triton X-114-derived OMPs [19]. We have also validated the expression of 8 proteins assigned as hypothetical by genome annotation, that are absent in the saprophytic non-pathogenic L. biflexa, a data that may implicate them in leptospiral pathogenesis.
In conclusion, we report for the first time the proteomic analysis of whole-cell, low-passage virulent strain, L. interrogans serovar Pomona sub-cultured from kidney and liver of infected hamsters. We have analyzed 895 spots and identified 286 of them as leptospiral proteins that corresponded to 108 different proteins - the highest number of proteins recognized by proteomics to date [15, 16, 18, 19]. These proteins are assigned in all functional categories and allocated in all bacterial cell compartments. We have validated the expression of 27 novel hypothetical leptospiral proteins, 5 of them containing SpI or SpII signal peptide cleavage sites and 8 that are absent in the non-pathogenic L. biflexa. We trust that, the global snapshot of proteome of L. interrogans expressed by virulent strain presented in this study would help to identify proteins of importance in pathogenesis, and hence, potential antigens for the development of vaccine against leptospirosis.
SUPPORTING INFORMATION
Supporting information can be viewed at www.bentham.org/open/tomicroj
ACKNOWLEDGMENTS
This work was supported by FAPESP, CNPq, and Fundação Butantan, Brazil; M.L.V. holds an MSc fellowship from FAPESP (Brazil).

