All published articles of this journal are available on ScienceDirect.

Ectopic Expression of Arabidopsis Phospholipase A Genes Elucidates Role of Phospholipase Bs in S. cerevisiae Cells
Abstract
In S. cerevisiae neither disruption of the phospholipase B triple knockout mutant (plb1plb2plb3; plb123) nor over-expression of phospholipase Bs (PLBs) result in a phenotype different from wild type. In performing experiments to characterize candidate plant phospholipase (PLA) genes, we found, surprisingly, that ectopic expression of either of two different A. thaliana PLA2 or PLA1 genes in the yeast plb123 mutant completely inhibited cell growth. We proposed that while PLBs might not be essential for growth and metabolism of yeast cells, they may play an important role in cell survival by metabolizing excess intracellular lysophospholipids. To test our hypothesis, we overexpressed a plant phospholipase A2 (PLA2) in both WT and plb123 cells, producing a pool of lysophosphatidylcholine (lysoPtdCho) in both transformants. In 14C acetate labeling experiments, WT cells were able to catabolize the resultant labeled lysoPtdCho, preventing accumulation, and the cells grew normally. In contrast, in the triple plb123 mutant PLA2 transformant, lysoPtDCho accumulated more than 4-fold to a toxic level, inhibiting cell growth. However, this growth inhibition was complemented by co-expression of either PLB1, PLB2 or PLB3 in the plb123 triple mutant already expressing the plant PLA2. Furthermore, in labeling experiments, the rescued cells exhibited a 60-75% reduction in 14C-lysoPtdCho build-up compared to plb123PLA2 cells. Our data provides conclusive evidence that yeast PLBs can metabolize intracellular lysoPtdCho produced by plant PLA2 overexpression in yeast. Our experiments indicate the utility of ectopic plant phospholipase A gene expression to characterize poorly-understood phospholipid metabolism mutants in yeast or other organisms.
INTRODUCTION
Phospholipids are primary components of all cell membranes. The main glycerolphospholipids in S. cerevisiae cells include, from most to least abundant, phosphatidylcholine (PtdCho), phosphatidylethanolamine (PtdEtn), phosphatidylinositol (PtdIns), phosphatidylserine (PtdSer) and phosphatidic acid (PtdOH). Membrane homeostasis is a dynamic condition wherein lipid synthesis, trafficking and degradation are coordinated to provide and sustain the integrity of different membranous subcellular structures [1]. In S. cerevisiae, lipid biosynthesis, its regulation and degradation are quite well understood, but much less is known about the biological function of lipid turnover, as well as the molecular mechanisms that coordinate those processes leading to membrane equilibrium.
As phospholipid turnover products, lysophospholipids (LysoPLs) have key roles in cell homeostasis. Due to their amphipathic nature, LysoPLs have surfactant and detergent-like properties. As a result, control of the in vivo levels of LysoPLs is essential for the function and survival of cells. In yeast biological membranes, LysoPLs are typically found at very low concentrations, approximately 0.02 to 0.08% of dry cell biomass, as derived from data of Blagovic et al. [2] and Summers et al. [3], respectively. The total lipid content of yeast varies widely with the species. The majority of yeasts contain 7%-15% total lipids as a proportion of dry weight [4]. Accordingly, the proportion of LysoPLs would fall into a range from 0.1%-1.1% of total lipids in different species. At low non-toxic concentrations, lysophospholipids which are metabolites of the mitogenically-responsive enzyme phospholipase A2, may be a mechanism for modulating the activity of downstream effector molecules. Lysophospholipids also interact with Ras proteins, regulatory elements for adenylyl cyclase, which catalyze the synthesis of cAMP, a secondary messenger critical for cell cycle progression [5]. However, higher concentrations of LysoPLs have been shown to disturb membrane conformation, affecting the activities of many membrane-bound enzymes, and may even lead to cell lysis and death [6]. As reported by Ko et al. [7], yeast (1,3)-β-glucan synthase was shown to be inhibited by phospholipase A2 and its reaction products, principally lysophospholipids; the inhibition was due, in part, to non-specific detergent-like membrane disruption. This may partially explain the resultant inhibition of cell multiplication. In wild type yeast cells, a typical phospholipid profile analysis does not show any appreciable lyso-phospholipid content [8]. In general, elevated LysoPL levels are usually characteristic of altered membrane phospholipid metabolism and impaired membrane integrity in particular.
In yeast, the de-acylation of PtDCho to produce glycerophosphodiester and FFA is currently known to be catalyzed by the enzymes Phospholipase B1, B2, B3 (Plb1p, Plb2p, Plb3p) and neuropathy target esterase (Nte1p), encoded by PLB1, PLB2, PLB3 and Nte1p genes, respectively. The PLB genes are highly similar (>60% identity at the DNA level). Phospholipase B activity catalyzes the deacylation of glycerophospholipids at both sn-1 and sn-2 positions. Plbps are located at the plasma membrane and in the periplasmic space with different properties [9]. Plb1p is the main enzyme responsible for PtdCho deacylation with its product, glycerophosphocholine (GroPCho), released into the extracellular medium. At their pH optima, substrate preferences of Plb1p and Plb2p are similar; both appear to accept all major phospholipid classes, whereas Plb3p accepts only PtdIns and PtdSer as substrates. At the physiological pH of yeast growth medium (around pH 5), Plb1p is primarily responsible for the degradation of PtdCho, and to some extent, PtdIns. Interestingly, overexpression of Plb2p does not result in degradation of PtdCho or PtdIns, while Plb3p hydrolyses PtdIns, but not PtdCho, in agreement with the substrate selectivity observed in vitro [9, 10]. Nte1p is a phospholipase B which resides in the endoplasmic reticulum, and prefers to deacylate PtdCho produced through the CDP-choline (Kennedy) pathway [11]. PtdCho can also be hydrolyzed by phospholipase D (Spo14p) to produce phosphatidic acid (PA) [12].
Both the triple PLB knockout mutant (plb123) and nte1 null mutant (nte1) strains are viable and do not exhibit any detectable phenotype beyond the lack of GroPCho formation [10]. It was reported that while the plb2 mutant and plb1plb2 double mutant grew at slightly reduced rates compared with wild type cells when plated on complete YEPD media [9, 10], the plb3 mutant and plb1plb3 double mutant and the plb1plb2plb3 triple mutant grew the same as wild type on fermentable and non-fermentable carbon sources [9, 10]. Merkel et al. [9] suggested that this may be due to the presence of alternative pathways, as was indicated by some residual release of GroPIns and GroPCho from a strain lacking all three phospholipase Bs. In fact, the plb123 mutant grows even better than wild-type cells [9; the current study), prompting questions about the possible physiological role of Plbps.
S. cerevisiae has proven to be a valuable experimental system to express and identify the function of plant genes involved in lipid metabolism. Because the plb123 triple mutant cells have a low background phospholipid deacylation activity, we initially tried to use the mutant as a system for screening and expressing some candidate plant phospholipase A (PLA) genes. Surprisingly, we found that overexpressing some of the putative PLA genes in plb123 completely inhibited the triple mutant cell growth, which suggested an important physiological role for PLBs. It had been reported that overexpression of PLB2 conferred resistance to the toxic effects of exogenous lysophosphatidylcholine (lysoPtdCho) supplied in the medium at a concentration of 10 mM (ca 0.5% w/v based on the MW of sn-1 palmitoyl LPC), which was thought to be consistent with its function as an extracellular lysophospholipase [8, 9]. Considering this result, the fact that Plbsp are located in plasma membranes, and because of the existence of other PtdCho-hydrolysing pathways (like that involving Nte1p), we proposed that Plbps, while perhaps not essential for metabolism and growth of yeast, may also play an important role in cell survival by catabolizing excess intracellular lysophospholipids. In order to test our hypothesis, we decided to reverse conventional approaches and instead of using yeast as a vehicle to characterize plant genes, use a known plant phospholipase A2 (PLA2), to collect relevant metabolic information on the poorly-understood yeast plb123 mutant. Theoretically, overexpressing plant PLA2 in yeast cells will produce a pool of lysophospholipids. If the cells posses the capacity to catabolize these lysophospholipids, preventing accumulation, the cells will grow normally. In contrast, if the cells are devoid of this capacity, lysophospholipids will accumulate to a toxic concentration. This test system provided direct proof that Plbps, as opposed to other pathways, can scavenge intracellular lysoPtdCho produced by overexpression of PLA2. Moreover, it indicated the utility of using plant PLA genes to screen yeast phospholipid mutants and understand nature of the lesions therein.
MATERIALS AND METHODOLOGY
General Molecular Biology Materials, Media, Radiochemicals and Lipid Analysis Supplies
Yeast extract, peptone, and agar were purchased from Difco® Laboratories, Detroit, MI. D-(+)-Glucose and D-(+)-galactose were acquired from Sigma-Aldrich Chemical Co., St. Louis, MO. Yeast nitrogen base without amino acids was a product of Becton, Dickinson and Company Spark, MD. Brent supplement mix (BSM) -URA was purchased from Sunrise Science, while BSM -LEU-URA was purchased from Formedium Ltd. Hunstanton, England. Escherichia coli strains Top10 and Topo10F’ competent cells, superscript II reverse transcriptase, DNA polymerase, pYES2.1 yeast shuttle vector, pCR®2.1-Topo® TA vector, S.c. EasyCompTm Transformation kit, restriction enzymes were purchased from Invitrogen, Inc., Carlsbads, CA. Pfu Turbo® DNA polymerase, pESC-Leu yeast vector were obtained from Stratagene. RNeasy® plant Mini Kit, QIA prep® Spin Miniprep Kit, QIA EX® II Gel Extraction Kit were products of Qiagen Inc., Mississauga, ON. L-α -1-palmitoyl-2-oleoyl [1-14C]-PtdCho was obtained from American Radiolabeled Chemicals, Inc, St, Louis. [1-14C]acetate (sodium salt) was purchased from Amersham Biosciences. Silica Gel 60 thin layer chromatography plates were obtained from Whatman Ltd., Maidstone, England. Oligonucleotide primers were synthesized by the Plant Biotechnology Institute. Organic solvents (HPLC grade or higher) were from Fisher Scientific or EMD Chemicals Inc., Damstadt, Gemany.
Yeast Strains, Growth Conditions and Transformation
All yeast strains used in this study (Table 1) were derivatives of the plb123 (plb1plb2plb3) triple mutant and its wild type, W3031A, kindly provided by Dr. Olaf Merkel. Synthetic minimal medium (SC) supplemented with appropriate nutrients (BSM-URA or BSM-LEU-URA plus glucose or galactose) were employed to select colonies containing various different plasmids. Yeast transformation was performed using the S.c. EasyCompTm Transformation kit. Genomic yeast DNA was isolated and prepared for gene cloning by the method of Sambrook and Russell [13]. Escherichia coli strains Top10 and Top10F’ were cultured in Luria broth and used for plasmid transformation by a standard method [14].
S.cerevisiae Strains Used in This Study
| Strains | Relevant Genotype | Genotype | Source |
|---|---|---|---|
| W3031A | PLB1PLB2PLB3 | Mata ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 | Olaf Merkel |
| MF17 | plb1plb2plb3 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX |
Olaf Merkel |
| WTEVa |
PLB1PLB2PLB3 YESpEV |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 YESpEV |
This study |
| WTPLA2 |
PLB1PLB2PLB3 PLA2 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 trp1–1 ura3–1 YESpPLA2 |
This study |
| plbEV |
plb1plb2plb3 YESpEV |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpEV |
This study |
| plbPLA2 |
plb1plb2plb3 PLA2 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpPLA2 |
This study |
| plbEVEV | plb1plb2plb3 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpEV ESCpEV |
This study |
| plbPLA2 EV |
plb1plb2plb3 PLA2 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpPLA2 ESCpEV |
This study |
| plbPLA2 PLB1 |
plb1plb2plb3 PLA2PLB1 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpPLA2 ESCpPLB1 |
This study |
| plbPLA2 PLB2 |
plb1plb2plb3 PLA2PLB2 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpPLA2 ESCpPLB2 |
This study |
| plbPLA2 PLB3 |
plb1plb2plb3 PLA2PLB3 |
Mata ade2–1 can1–100 his3–11,15 leu2–3,112 ura3–1 plb1::TRP plb2::HIS plb3::kanMX YESpPLA2 ESCpPLB3 |
This study |
a EV is short for empty vector.
Primers Used in This Study. NotI or SacI Sites are Underlined
| Primer Name | Primer Sequence |
|---|---|
| PLA2F87 | ACTATGGCGGCTCCGATCA |
| PLA2R536 | TTAGGGTTTCTTGAGGACTTTGC |
| ACTF | ATGGATTCTGAGGTTGCTGCTT |
| ACTR | AATGGCGTGAGGTAGAGAGAAAC |
| PLB1F1 | GCGGCCGCATGAAGTTGCAGAGTTTGTTGG |
| PLB1R1995 | GAGCTCCTAAATTAGACCGAAGACGGC |
| PLB2F1 | GCGGCCGCATGCAATTACGGAACATATTACAG |
| PLB2R2121 | GAGCTCCTAAATTAGTCCAAATAATGCTGTTAT |
| PLB3F1 | GCGGCCGCATGATACGTCCATTATGTTCAAAA |
| PLB3R2061 | GAGCTCTTATACTGCTCCGGTAAACATCA |
The Growth Inhibition of the plb123 Mutant Upon AtPLA2 Expression is Principally the Result of Intracellular Accumulation of lysoPC. In Co-Cultivation Experiments, the plb123 Mutant was Transformed by Empty Vectors pYES2.1 and pESC with URA and LEU Selection Markers (Called plbEVEV), WT was Transformed by the Empty Vector (Called WTEV) or by PLA2 (Called WTPLA2) with the URA Selection Marker. The WTPLA2 was Then Co-Cultured with plbEVEV (WTEV Co-Cultured with plbEVEV was Used as the Control). Cells were Induced to Determine Whether WTPLA2 Could Release Enough lysoPC to Affect the Growth of plb123 Mutant Cells. In Order to Fully Reflect the PLA2 Effect, 10-Times the Number of WTPLA2 Cells and WTEV Cells were Grown Against plbEVEV Cells, which were Mixed Together for Co-Culture as Follows: After WTEV, WTPLA2 and plbEVEV were Cultured to OD600 3.0, WTEV and WTPLA2 Cells were Each Diluted to OD600 1.0 while plbEVEV Cells were Diluted to OD600 0.1 and Then the Preparations Co-Cultured. Then Either Before Induction or After Two Days Induction in the Presence of Galactose, Cultures were Diluted 100-fold and 10 µl was Spread on Plates with SC-URA-LEU Medium and Colonies Counted.
| Co-Culture Strain Combination | Induction Time | # of Cells / mL Culture |
|---|---|---|
| WTEV + plbEVEV | 0 hr | 2.77E+05 ± 1.533E+04 |
| WTPLA2 + plbEVEV | 0 hr | 2.83E+05 ± 5.774E+03 |
| WTEV + plbEVEV | 48 hr | 1.40E+06 ± 1.447E+05 |
| WTPLA2 + plbEVEV | 48 hr | 1.42E+06 ± 1.422E+05 |
Values are reported as # of colonies per mL of culture and are the average of three replicates ± SD.
Cloning the PLA2 from Arabidopsis and Expression in Yeast
Total RNA was extracted from Arabidopsis leaves using the RNeasy® plant Mini Kit and cDNA was synthesized by superscript II® reverse transcription according to the product instructions. The primers PLA2F87 and PLA2R536 (see Table 2) were designed to amply the coding region of phospholipase A2 (At2g06925) [14], designated as PLA2. Arabidopsis leaf cDNA was used to amplify PLA2 by Pfu Turbo® DNA polymerase. To the resultant blunt end PCR product, an adenine overhang was added at the 3' end using Taq DNA polymerase, and the amplicon cloned into the yeast shuttle vector pYES2.1 in the Top 10F’ strain. After confirming cloning integrity by sequencing, the vector was transformed into both the WT and the plb123 triple mutant yeast strains using the S.c. EasyCompTm Transformation kit. The self-ligated pYES2.1 was also transformed into the triple mutant and WT strains as controls (see Table 1).
RT-PCR Confirmation of PLA2 Expression in WT Yeast
Both wild-type S. cerevisiae bearing the empty vector and yeast cells bearing PLA2 were inoculated in YPD medium containing 2% of dextrose. Cells were cultured at 28°C for 24 hr, followed by culture in YPD medium containing 2% β-galactose at 28°C for 12 hr. Total RNA was isolated from both wild-type S. cerevisiae bearing the empty vector and transgenic cells bearing PLA2 using the standard method of Sambrook and Russell [13]. To examine the transcript levels in transformed yeast, two micrograms of DNase I-treated RNA were used as a template for cDNA synthesis (Invitrogen superscript First-Strand Synthesis kit). To exclude genomic DNA contamination, DNA-free RNAs were tested by PCR using pLA2 primers (See Table 2). The yeast actin gene ACT1/YFL039C was amplified (using primers ACTF and ACTR, Table 2) and used as an endogenous standard to confirm mRNA quality. The cDNA samples were diluted 50-fold for PCR amplification. The PCR program: DNA denaturation at 94°C for 4 min, followed by 35 cycles of PCR at 94°C for 30 seconds; primer annealing at 53°C for 30 seconds and extension and incubation at 72°C for 5.75 min, then cooling at 4°C. PCR products were resolved by electrophoresis on 1% agarose gels. The expected amplified fragment size of yeast actin and pLA2 are 525 bp and 450 bp, respectively. Functional expression of the PLA2 was confirmed by phospholipase A2 assay as described below.
Plate Assay of Strain Growth
All strains were cultured to stationary phase in SC medium with glucose and adjusted to an OD600 of 3.0 as a starting concentration; cells were also diluted 1:10, 1:100 and 1:1000 with SC medium without saccharide. Ten μl of each was spotted on plates with different media containing either glucose or galactose. After 2, 3 and 5 days, the degree of cell growth was determined.
Phospholipase A2 Assay
PLA2 activity was measured as the release of radioactive free [1-14C] oleic acid from the substrate L-α-1-palmitoyl-2-oleoyl[1-14C]-PtdCho. The substrate was dried under a stream of N2 before adding the other reaction components. Standard enzyme assay mixtures contained 50 mM Tris-HCl, pH 8.0, 10 mM CaCl2, 0.05% (v/v) Triton X-100, 0.1μCi of the substrate (55mCi/mmol), in a total volume of 200 μl. The mixtures were emulsified by sonication at room temperature for 3 min. and reactions started by addition of 60 μg of transformant lysate protein. The reactions were incubated in a shaking water bath at 30ºC for 30 min and terminated by the addition of 750 μL of chloroform: methanol (1:2, v/v). The lipids were extracted and spotted onto a thin layer chromatography plate (Silica Gel G), which was developed with chloroform: methanol: NH4OH: water (73:30:4:1). The spots corresponding to the lipid standards FFA and lysoPtdCho, were then scraped into vials, 4 ml scintillation fluid added (Aquasol-2, DuPont) and the radioactivity determined by standard scintillation counting.
Labeling Yeast Cells with Radioactive Sodium Acetate and Lyso-Phosphocholine Analysis
Yeast cells were cultured in SC-URA medium with glucose. The cells from stationary stage cultures were diluted to OD600 0.1 in a 20 ml volume. 30µCi of [1-14C]- labeled sodium acetate was diluted in SC medium and the labeled solution filtered before adding to the cultures. The cells were inoculated and incubated for 10 hours at 28ºC. The cultures were then divided in half; One half continued to be incubated in glucose medium, while with the other half, collected cells were washed twice with sterilized water and transferred to galactose medium. After 5 hours incubation, cells were collected and washed twice with PBS (10 mM phosphate, 2.7 mM KCl, 137 mM NaCl, pH7.4), to be analyzed for lipids. Cells were homogenized with an equal volume of glass beads using the BeadBeater at 4ºC using 3 bursts of 1 min. Total lipids were extracted by adding 2 ml of chloroform:methanol:KCl/HNO3(1:2:0.8,v/v/v) vortexing and then adding 2ml dichloromethane and the total lipids were recovered from the lower organic phase. The labeled lipids were separated by two-dimensional thin layer chromatography on Silica gel 60 plates using chloroform/methanol/ acetic acid/water (80:9:12:2) as the first, and chloroform/methanol/acetic acid/water (50:25:10:2) as the second developing solvent, respectively. The TLC plates were screened by a TLC Imaging Scanner (Bioscan, WA, USA). Relevant spots were scraped and quantified by liquid scintillation counting.
Co-Culture Experiments
The plb123 mutant was transformed by two empty vectors with URA and LEU selection markers respectively (designated plbEVEV). while the wild type cells were transformed by PLA2 expression vector or its empty vector pYES2.1 with URA selection marker only and designated as WTPLA2 and WTEV, respectively. The WTPLA2 was then co-cultured with plbEVEV (WTEV co-cultured with plbEVEV was used as the control). Under the induced condition, we wanted to determine whether WTPLA2 could release enough lysoPC to affect the growth of plb123 mutant cells. In order to fully reflect the PLA2 effect, 10-times the number of WTPLA2 cells and WTEV cells were grown against plbEVEV cells, which were mixed together for co-culture as follows: After WTEV, WTPLA2 and plbEVEV were cultured to OD600 3.0, WTEV and WTPLA2 cells were each diluted to OD600 1.0 while plbEVEV cells were diluted to OD600 0.1 and then the preparations co-cultured. Then either before induction or after two days induction in the presence of galactose, cultures were diluted 100-fold and 10µl was spread on plates with SC-URA-LEU medium and colonies counted.
Cloning the PLB1, PLB2 and PLB3 Genes from Yeast and Expression in the plb123 triple Mutant
Yeast DNA was extracted by the method of Sambrook and Russell [13]. The primers PLB1F1, PLB1R1995, PLB2F1, PLB2R2121, PLB3F1, PLB3R2061 (see Table 2) with NotI or SacI sites (underlined) were designed to amply the coding region of PLB1, PLB2, and PLB3, respectively using Pfu Turbo® DNA polymerase. To the resultant blunt end PCR products, adenine overhangs were added at the 3' ends using Taq DNA polymerase and the amplicons cloned into pCR®2.1-Topo® TA vector. After confirming sequence integrity, the plasmids were digested by NotI/SacI, the 1.99kb PLB1, 2.12kb PLB2 and 2.06kb PLB3 were recovered from the gel and each ligated into the NotI/SacI sites in the yeast shuttle vector pESC-LEU. The plasmids and the pYES2.1PLA2 (described above) were co-transformed into the plb123 triple mutant using the S.c. EasyCompTm Transformation kit and transformant cells selected on SC-LEU-URA medium. The self-ligated pYES2.1 and empty pESC-Leu vector were also used to transform the triple mutant strain as a control. The product strains and their genotypes are shown in Table 1.
RESULTS
While screening a number of putative plant PLA genes by over-expression in the plb123 triple mutant strain, we found that there was generally a complete inhibition of growth. Thus, we considered using a reverse strategy- one of using a known plant PLA2 gene to determine whether PLBs may play an important role in cell survival, perhaps by scavenging excess lysophospholipids. Although there are reports that PLA activity can be measured in yeast, there have been no yeast PLA genes cloned thus far [15], making the availability of the plant PLA gene even more valuable in probing yeast phospholipid metabolism mutations. A PLA2 gene from Arabidopsis (At2g06925) encodes a 149-amino acid protein, identified as phospholipase A2, which deacylates phospholipids at the sn-2 position to produce lysophospholipids and FFAs [14]. The Arabidopsis PLA2 was cloned into the yeast shuttle vector pYES2.1, in which PLA2 was driven by GAL1 promoter (see Materials and methods). The GAL1 promoter can be inhibited by glucose and stimulated by galactose [16].
Functional Expression of the Arabidopsis PLA2 in Yeast
WT yeast strain W3031A was transformed with either pYESPLA2 plasmid or with empty vector (control). PLA2 expression was confirmed by RT-PCR (Fig. 1). To assess whether the expressed AtPLA2 had functional activity, cells were collected from exponential stage cultures induced in galactose medium, lysed and the lysate used in phospholipase A2 assays as described in Materials and methodology. The release of sn-2 14C-labeled oleic acid from L-α-1-palmitoyl-2-oleoyl[1-14C]-PtdCho was 15-fold higher than the low background activity in the WT yeast. This experiment confirmed that the plant PLA2 gene was functionally expressed in yeast cells and encoded a protein with phospholipase A2 activity. Furthermore, expression of the AtPLA2 was not toxic to WT yeast.
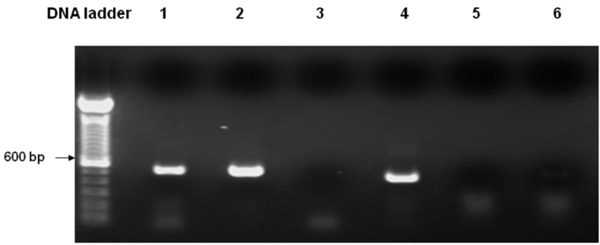
Expression of Arabidopsis PLA2 in WT yeast as determined by RT-PCR. Lane 1. Template: WTEV cDNA, Actin primers; Lane 2. Template: WTPLA2 cDNA, Actin primers; Lane 3. Template: WTEV cDNA, PLA2 primers; Lane 4. Template: WTPLA2 cDNA, PLA2 primers; Lane 5. Template: DNA-free RNA from WTEV, PLA2 primers; Lane 6. Template: DNA-free RNA from WTPLA2, PLA2 primers. Lanes 1 & 2 show that the cDNA from both empty vector- and PLA2- transformed yeast were of good quality, as the yeast actin was constitutively expressed in both samples. Lane 3: The same empty vector-transformed WT yeast as in Lane 1, failed to produce the PLA2 transcript; Lane 4: In contrast, PLA2-transformed WT yeast produced the expected 450 bp PLA2 transcript. For Lanes 5 & 6, using DNA-free RNA as the template there is no signal with the PLA2 primers, confirming that there is no possibility that genomic DNA contamination in the cDNA templates could have resulted in the PLA2 band as observed in Lane 4.

Cell growth of Wild type empty vector (WTEV), wild type PLA2 (WTPLA2), plb123 empty vector (plbEV) and plb123 PLA2 (plbPLA2) transformants (as designated in Table 1) on medium supplemented with glucose or galactose. All strains were cultured to stationary phase in SC medium with glucose and adjusted to OD600 of 3.0 as a starting cell density; Cells were also diluted 10, 100 and 1000 times and 10 μl of each was spotted on a plate with medium as indicated and incubated for three days.
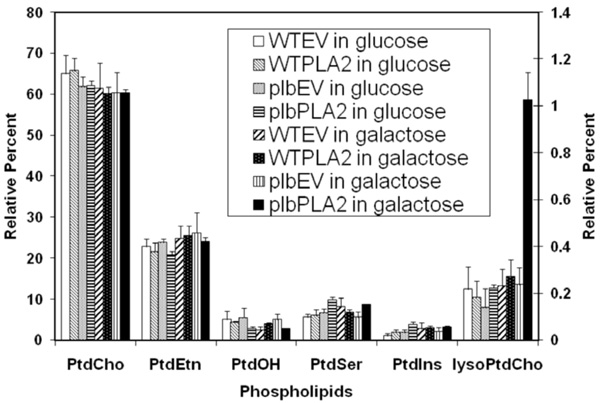
Relative 14C labeling of various phospholipids isolated from transformed strains fed with 14C acetate. Starting from a cell density of 0.1 (OD600) in 20 ml of glucose medium, cells of WTEV, WTPLA2, plbEV and plbPLA2 (as designated in Table 1) were labeled with 30µCi 14C-sodium acetate for 10 hours. Half of culture remained in glucose medium while the other half was induced in galactose medium for 5 hours. The total lipids were extracted and resolved by two dimensional thin layer chromatography and each of the phospholipids was quantified (see Materials and methodology). The proportions of 14C-labeled LysoPtdCho (relative to the total 14C phospholipids) are depicted using the Y axis scale on the right side, while the proportions of other 14C-labeled phospholipids are expressed using the Y axis scale on the left. The results are the means ±S.E of three experiments.

Co-expression of either PLB1 or PLB2 or PLB3 with PLA2, respectively, in the plb123 triple mutant. All strains are designated by those plasmids expressed in the plb strain as designated in Table 1 (e.g. plbPLA2PLB1 for the plb triple mutant co-expressing the Arabidopsis PLA2 and the yeast PLB1). Transformants were cultured to the stationary phase in SC medium with glucose and adjusted to an OD600 of 3.0 and also diluted by 10, 100 and 1000-fold; 10 µl of each was spotted on plates with different media. The photograph was taken after 3 days of growth.
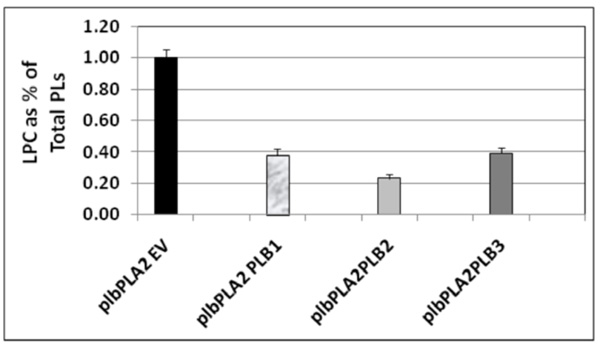
Relative 14C lysoPtdCho content in 14C acetate labeling experiments conducted following co-expression of either PLB1 or PLB2 or PLB3 with PLA2, respectively, in the plb123 triple mutant. All strains are designated by those plasmids expressed in the plb strain as explained for Table 3 and indicated in Table 1. Starting from a cell density of 0.1 (OD600) in 20 ml of glucose medium, cells of plbPLA2EV, plbPLA2PLB1, plbPLA2PLB2 and plbPLA2PLB2 were labeled with 30 µCi 14C-sodium acetate for 10 hours, then induced in galactose medium for 5 hours. The total lipids were extracted and resolved by two dimensional thin layer chromatography and the 14C lysoPtdCho was quantified (see Materials and methods). The plb123 triple mutant expressing only the PLA2, designated as plbPLA2EV, accumulated 14C lysoPtdCho to 1.0 % of the total phospholipid (PL) fraction, as was typical in previous experiments (See Fig. 3). The plbPLA2PLB1, plbPLA2PLB2 and plbPLA2PLB3 co-transformants exhibited 60-75% reductions in the relative 14C lysoPtdCho content. Values are the average of 2 replicates.

Simplified scheme of lysoPtdCho metabolism in Saccharomyces cerevisiae. Abbreviations are:LysoPtDCho (lysophosphatidylcholine), GroPCho (glycerophosphocholine), PtDCho (phosphatidylcholine), LPCAT (lysophosphylcholine:acyl-CoA acyltransferase), mtTaz1 (mitochondrial acyl-CoA-independent LPCAT; [25]; Plb1p, Plb2p, Plb3p (phopholipases B1, B2 and B3), Nte1p (neuropathy target esterase); Dnf1p and Dnf2p (aminophospholipid translocating P-type ATPases 1 and 2, respectively); Lem3p (noncatalytic β subunit of Dnf1p and Dnf2p); GPCAT (glycerophosphocholine acyltransferase); LRO1 (phospholipid:diacylglycerol acyltransferase; PDAT; [19, 26]; Git1P (glycerophosphoinositol/glycerophosphocholine transporter; [27]. Not shown in schematic: PLBs are also involved in the hydrolysis of PC and PI to produce extracellular glycerophosphocholine and glycerophophoinositol, respectively [28].
The Over-Expression of Plant PLA2s Completely Inhibits the Growth of Yeast plb123 Triple Mutant Cells but has no Obvious Effect on Wild Type Cells
When the Arabidopsis PLA2 was introduced into the plb123 triple mutant and maintained on SC-URA medium with glucose, the PLA2 was not expressed, and there were no obvious changes in cell growth compared to the empty vector controls or the WTPLA2 transformants. Once cultured in the medium containing galactose, the empty vector control cells and WTPLA2 cells grew normally, but the growth of plb123 cells harbouring PLA2 (plbPLA2) was completely inhibited (Fig. 2). The results were identical even after 5 days of incubation (data not shown). These results show that the growth inhibition of the plb123 triple mutant was due to the expression of PLA2, and that such inhibition acts only in the plb123 triple mutant cells, but not in wild type cells.
To determine whether the cells were dead or growth merely inhibited when PLA2 was expressed, cells induced for 3 and 6 days in galactose liquid medium were collected and washed, re-suspended in glucose medium and re-plated on glucose plates. The cells were once again able to grow, suggesting that overexpression of AtPLA2 was not lethal to the plb123 cells, but rather, temporarily inhibited cell growth (data not shown).
The Over-Expression of Plant PLA2s Leads to A Toxic Accumulation of lysoPtdCho in plb123 Cells
Once we confirmed that the plant PLA2 was functionally expressed (see above), we speculated that its overexpression could be used to stimulate an accumulation of lysophospholipids and/or free fatty acids, which might be toxic to the plb123 cells. No obvious accumulation of free fatty acids was found in growth-inhibited cells (data not shown). Because of the very low level of endogenous lysophospholipids, such as lysoPtdCho, in yeast cells, it was necessary to label the phospholipids by feeding cells with 14C acetate to estimate relative polar lipid pool sizes. In preliminary experiments, we found that the phospholipids were well-labeled during the exponential phase but not in later phases (data not shown). This is consistent with the fact that neutral, not polar, lipids accumulate in the late phase of cell growth. Thus labeling was performed during the exponential phase of growth.
While most major phospholipids of WTEV, WTPLA2, plbEV and plbPLA2 strains were all labeled, among lyso phospholipids only lysoPtdCho (and not lysoPtdEtn, lyso PtdIns, lysoPtdSer) was significantly labeled. The level of lysoPtdCho in induced plbPLA2 increased more than 4-fold over the low endogenous level consistently observed in control cells (Fig. 3). In WT cells expressing PLA2 (WTPLA2) there was no enhanced accumulation of lysoPtdCho and cell growth was unaffected. Collectively, these results mean that the over-expression of plant PLA2 can be used to produce lysophospholipids, mainly lysoPtdCho, which can be catabolized by wild type yeast, but accumulates in the plb triple mutant. This suggests that phospholipase Bs play an important role in scavenging excessive lysoPtdCho in yeast cells and that the plb123 mutant has lost this ability.
The Growth Inhibition of the plb123 Mutant Upon AtPLA2 Expression is Principally the Result of Intracellular Accumulation of lysoPC
Because in Arabidopsis, PLA2, has been observed in both intracellular and extracellular spaces [14] , the possibility remained that the expressed AtPLA2 is producing lysoPC extracellularly at a level high enough to also inhibit the growth of the plb123 mutant. In that case, using PLA2 expression to produce lysoPC would not be a qualitatively different experiment than providing lysoPC to the culture media. To demonstrate that it was the ectopic expression of the plant PLA2 giving rise to increased intracellular lysoPC that was primarily responsible for the growth inhibition, we designed the following co-culture experiment: The plb123 mutant was transformed by two empty vectors with URA and LEU selection markers, respectively (designated plbEVEV), while the wild type cells were transformed with the PLA2 expression vector or with its empty vector pYES2.1, with URA selection marker only, and designated as WTPLA2 and WTEV, respectively. We then co-cultured WTPLA2 with plbEVEV and, as a control, we co-cultured WTEV with plbEVEV. Under induced (+ gal) conditions, we wondered whether WTPLA2 might release some amount of extracellular lysoPC, which might affect the growth of the plb123 mutant cells. After two days of induction, the co-cultures were diluted and spread on plates with SC-URA-LEU medium. Due to the presence of the both selection nutrients, only plbEVEV cells were able to grow, while WTEV or WTPLA2 could not. By counting colony numbers it was possible to evaluate the effect of lysophospholipids released by WTPLA2 on plbEVEV cells. Our results showed that there is no difference in growth between the two co-cultures (Table 3). Combining these results with those from 14C acetate labeling experiments (above), it is clear that the intracellular accumulation of lysoPC by AtPLA2 expression was the principle reason for the growth inhibition of the plb123 mutant.
The Expression of PLB1, or PLB2 or PLB3 can Eliminate the Inhibition of PLA2s on plb123 Triple Mutant Cell Growth
In order to determine whether one or other of the phospholipase Bs could rescue plb123 cell growth which had been inhibited by the over-expression of AtPLA2, each PLB gene was co-expressed separately with PLA2 in plb123. All three PLB genes could rescue cell growth (Fig. 4). Under our experimental conditions, there was no difference in the rate of growth in the rescued cells. Most importantly, 14C acetate labeling experiments showed that those cells rescued by co-expression of any of the three PLB genes exhibited a 60-75% reduction in the 14C lysoPtdCho which otherwise accumulated to a toxic level in the case of the plb123PLA2 transformant (Fig. 5). Thus, it appears that lysoPtdCho threshold proportions of 0.5% or higher are highly toxic to the plb123 mutant, which is supported by the earlier findings of Merkel et al. [9] and Fyrst et al. [8] in studies of PLPB2 as a putative extra-cellular lysophospholipase. We also co-expressed in plb123, one of PLB1, PLB2 or PLB3 with another Arabidopsis PLA gene, PLA1 (At2g44810), and found that the growth inhibition caused by this PLA1 could also be rescued by each of the PLBs (data not shown). These results demonstrate that PLBs are redundant in scavenging excess lysoPtdCho, and that such mechanisms are generally induced by plant PLA expression in plb123 cells.
DISCUSSION
LysoPtdCho is an important lysophospholid in cells; its potential generation and degradation in yeast cells is summarized in the simplified model shown Fig. (6). The production of lysoPtdCho occurs via different pathways: One is via the deacylation of PtdCho by Plb1p, Plb2p and Nte1p [8, 9, 11, 17]; In the other pathway, lysoPtdCho is taken up into the intracellular compartment by the action of Dnf2p [18, 19]. When GroPcho is taken up into yeast by Git1p, lysoPtdCho can be produced by GPCAT [20, 21]. LysoPtdCho can be produced in vitro in yeast lysates via Lro1, transferring an acyl group from PC to diacylglycerol [22].
There are several ways to eliminate excess lysoPtdCho. LysoPtdCho can be converted to PtdCho in the presence of acyl-CoA by lysophosphylcholine:acyl-CoA acyltransferase (LPCAT; EC 2.3.1.23) and there are several recent reports to support this [23-27]. Over-expression of LPCAT can enhance the cell’s resistance to lysoPtdCho [27]. In yeast, there is also a mitochondrial acyl-CoA-independent LPCAT activity, mediated by Taz1 [28]. Finally, lysoPtdCho can also be catabolized via Plb1p, Plb2p, Plb3p or Nte1p. Overexpression of PLBs was known to enhance cell resistance to lysoPtdCho and the plb123 triple mutant cells showed more sensitivity to LysoPtdCho [8, 9] . At the time, extracellular PLB activity was known but the existence of the exogenous lysolipid metabolism (ELM) pathway, reported only recently [19], was not. Thus it was thought that PLBs could degrade exogenous lysophospholipids and increase the resistance to the occurrence of such lipids in the medium; the degradation of LysoPtdCho by PLBs was considered to occur only extracellularly.
In our hands, and consistent with these previous studies, when cultured on synthetic minimal medium (SC) supplemented with appropriate nutrients (BSM-URA or BSM-LEU-URA plus glucose or galactose), we did not observe any apparent difference in growth rate between the wild type and plb1plb2plb3 triple mutant. In this study, by means of expressing an AtPLA2 in plb123 and WT cells and comparing their growth with un-induced cells, we found that expression of AtPLA2 could completely inhibit growth of the plb123 triple mutant cells, but not that of wild type cells. Further investigation revealed that excessive lysoPtdCho accumulation occurred only in plb123 cells expressing the AtPLA2. Co-cultivation experiments supported the hypothesis that this effect was due primarily to intracellular, rather than extracellular, lysoPtdCho accumulation.
Complementation experiments showed that all three PLBs could rescue the growth inhibition of plb123 triple mutant cells caused by AtPLA2 expression. Finally, in addition to relief from growth inhibition, the cells of plb123PLA2 complemented with PLBs also exhibited a dramatic 60-75% reduction in lysoPtdCho accumulation. Collectively, these findings directly support our hypothesis that Plbps play a very important role in catabolizing excess intracellular lysoPtdCho. Phospholipase B exhibits three activities in vitro: 1) phospholipase A activity; 2) lysophospholipase activity; 3) transacylase activity. The lysophospholipase activity of Plb1p and Plb2p is about 100 fold higher than its PLPase A activity; even its transacylase activity is higher than that of PLPase A. In this regard, it is probably more suitable to refer to Plbs as lysophospholipases or transacylases, rather than phospholipase Bs. As the exception, no lysophospholipase activity has been detected in Plb3p, but it shows some transacylase activity [12]. Thus, our observation that Plb3p can rescue the growth inhibition of the plb123 triple mutant expressing the AtPLA2, might be due to Plb3p acting as a transacylase.
CONCLUSION
In conclusion, our results show that PLBs can efficiently eliminate excess lysoPtdCho produced by PLA2 to maintain healthy cell growth. In the absence of any experimental evidence, it cannot be excluded that extrusion and processing by transporters such as Nte1p, also contribute to the processing of lysoPC to maintain homeostasis. It is, however, interesting to note that, even though NTE1 and LPCAT are still present in the plb123 mutant, our results suggest that the intracellular lysoPtdCho which accumulates in the presence of overexpressed Arabidopsis PLAs is more reliant on PLBs to maintain yeast membrane homoestasis through lipid catabolism. Our results do not preclude other roles for PLBs. Given that the common reaction product of PLAs acting on PtdCho is lysoPtdCho, and that we were able only to observe increases in this specific lyso-phospholipid upon expression of PLA2, it is reasonable to assume that the over-expression product in this case is solely lysoPtdCho. However, this does not a priori, rule out other unknown lipid products being produced.
ACKNOWLEDGEMENTS
We wish to thank Dr. Olaf Merkel, who generously supplied the plb123 triple mutant and its wild type, W3031A. We also thank Drs. Pierre Fobert, Mark Smith and Jitao Zou for helpful discussion and suggestions during the course of this study. This is National Research Council of Canada Publication No. 50125.

