All published articles of this journal are available on ScienceDirect.

In Silico Design and Evaluation of a Novel Chimeric Vaccine Candidate Based on SAG1, GRA1, and MIC4 Antigens of Toxoplasma gondii
Abstract
Introduction/Background
Toxoplasma gondii (T. gondii) is an intracellular protozoan parasite that poses serious risks to immunocompromised individuals and pregnant women. The adverse effects of current anti-toxoplasmosis drugs highlight the need for an effective vaccine. This study aimed to design a novel chimeric vaccine composed of selected epitopes from SAG1, GRA1, and MIC4 antigens using immunoinformatics approaches.
Methods
Immunodominant B- and T-cell epitopes were predicted using the Immune Epitope Database (IEDB) and PRED (BALB/c) tools. Selected epitopes were linked via an A(EAAAK)nA linker to construct the SGM (SAG1-GRA1-MIC4) chimeric protein. Structural properties, physicochemical characteristics, antigenicity, allergenicity, and solubility were evaluated using online bioinformatics servers.
Results
The SGM construct consisted of 395 amino acids with a predicted molecular weight (MW) of 42.62 kDa and an isoelectric point (pI) of 5.53. Structural validation indicated favorable stereochemical quality, with 96.08% of residues in favored regions of the Ramachandran plot. The protein was predicted to be stable, soluble, antigenic, and non-allergenic. Codon optimization analysis suggested efficient expression potential in the selected host system.
Discussion
The integration of immunodominant epitopes from three major T. gondii antigens into a single construct may enhance immune coverage and vaccine efficacy. In silico analyses support the structural stability and immunogenic potential of the designed construct, suggesting its suitability as a multi-epitope vaccine candidate.
Conclusion
The SGM construct represents a promising multi-epitope vaccine candidate against T. gondii; however, experimental validation is required to confirm its immunoprotective efficacy.
1. INTRODUCTION
Toxoplasma gondii (T. gondii) is an important zoonotic protozoan parasite that can infect almost any warm-blooded animal, such as cows, sheep, goats, camels, and birds. It can also infect cold-blooded animals like frogs, toads, turtles, crocodiles, snakes, and fish [1-6]. Infection with this parasite is caused by consuming raw or undercooked meat containing tissue cysts or consuming food or drinking water contaminated with oocysts shed by cats. Furthermore, organ transplantation, blood transfusion, and vertical transmission from mother to fetus during pregnancy are other routes of T. gondii transmission [7, 8]. The life cycle of this parasite includes three stages: the tachyzoite, which is responsible for acute infection; the bradyzoite, which causes chronic infection; and the sporozoite, which is enclosed within the oocyst [9]. Infection is usually asymptomatic in people with healthy immune systems, but latent toxoplasmosis can lead to behavioral disorders in humans [10-19] that are associated with changes in the levels of neurotransmitters in the central nervous system [20, 21]. In contrast, immunocompromised patients face life-threatening complications, such as reactivation of chronic infection leading to toxoplasmic encephalitis [22]. Additionally, T. gondii acquired during pregnancy may lead to miscarriage, congenital birth defects, or chorioretinitis in the fetus [23].
Current therapies focus on the tachyzoite stage, and there are no effective options for eliminating tissue cysts [24]. This parasite is resistant mainly due to cysts that have protective walls and low metabolic activity, which allows it to evade the host's immune system and drugs [25, 26]. Moreover, conventional treatments, which include a combination of pyrimethamine and sulfadiazine, have many side effects, highlighting the need for safer preventive strategies [27]. Vaccine development represents a promising approach against toxoplasmosis in both humans and animals. Recently, peptide-based vaccines have gained attention due to their ability to induce potent humoral and cellular immune responses. In silico methods help to rapidly identify and design immunogenic T and B cell epitopes from important parasite antigens, accelerating vaccine development [28]. Surface antigens (SAGs) and secretory excretory antigens [dense granule proteins (GRAs), rhoptry proteins (ROPs), and microneme proteins (MICs)] play central roles in host–parasite interactions and are therefore attractive targets for vaccine development against T. gondii [29]. Among these, SAG1 is the major surface antigen of tachyzoites and is critically involved in host cell attachment and immune recognition, stimulating both humoral and cellular immune responses [30]. GRA1 is secreted into the parasitophorous vacuole and contributes to parasite survival and intracellular persistence [31]. MIC4 is involved in parasite motility, host cell recognition, and invasion, representing a key factor during the early stages of infection [32]. Previous vaccine studies using SAG1, GRA1, MIC4, or their combinations-mainly in full-length protein or DNA-based formats-have demonstrated partial protection and the induction of Th1-biased immune responses, highlighting their immunogenic potential while also underscoring the need for improved vaccine design strategies [29, 32-35]. Therefore, the present study aimed to design a chimeric protein vaccine by integrating immunodominant epitope-rich regions from SAG1, GRA1, and MIC4 using a rational immunoinformatics-based approach. Unlike previous multi-antigen vaccine studies, this work focuses on the selective enrichment of overlapping B- and T-cell epitopes to maximize protective immunogenicity while minimizing non-essential or potentially non-protective sequences.
2. MATERIALS AND METHODS
2.1. Retrieval of Protein Sequences
The amino acid sequence of SAG1, GRA1, and MIC4 antigens was obtained from the Universal Protein Resource (UniProt) (http://www.uniprot.org/) in FASTA format for the T. gondii RH strain. These sequences formed the basis of subsequent analyses performed using bioinformatics tools.
2.2. Prediction of Transmembrane Domains and the Signal Peptides
Transmembrane domains were predicted using the TMHMM Server version 2.0 (http://www.cbs.dtu.dk/ services/TMHMM-2.0) [36]. Proteins with an expected number of amino acids in transmembrane helices (Exp number of AAs in TMHs) greater than 18 were considered likely transmembrane proteins. Additionally, for the first 60 N-terminal amino acids, if the expected number exceeded 10, a possible N-terminal signal sequence was flagged. The total probability of the N-terminal being inside the cytoplasm (Total prob of N-in) was also considered. Predicted regions are reported as inside, outside, or transmembrane, with the orientation of the helices indicated.
Signal peptides were predicted using SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP) [37] with default parameters. Signal peptides were defined based on the D-score exceeding the cutoff value, and the predicted cleavage sites were recorded. Predicted signal peptide regions were excluded from epitope selection for recombinant protein design.
2.3. Prediction of B-cell Epitopes
Linear B-cell epitopes in SAG1, GRA1, and MIC4 were predicted using the Immune Epitope Database (IEDB) analysis resource (http://tools.iedb.org/bcell/). IEDB integrates multiple validated algorithms for B-cell epitope prediction, including linear epitope propensity, β-turns, flexibility, hydrophilicity, antigenicity, and surface accessibility [38–43]. Regions that consistently scored higher on these parameters were considered potential B-cell epitopes and selected for further analysis.
2.4. Major Histocompatibility Complex (MHC) Class I and II Binding Epitope Prediction
Potential T-cell epitopes were identified using the PRED (BALB/c) tool (http://cvc.dfci.harvard.edu/balbc/), which is designed for predicting MHC binding peptides for the BALB/c mouse model. Three MHC class I alleles (H2-Kd, H2-Ld, H2-Dd) and two MHC class II alleles (H2-IAd, H2-IEd) were assessed. Nine-amino-acid peptides were scored on a scale of 1–10 based on predicted binding affinity. Peptides with binding scores greater than 9 were considered high-affinity binders and selected for further analysis. Predictions were cross-validated using IEDB to increase reliability [44].
2.5. Construction of Fusion Peptides
Immunodominant B- and T-cell epitope-containing regions from SAG1, GRA1, and MIC4 antigens were linked to create chimeric constructs. A(EAAAK)nA linkers were used to connect the domains and promote stable, correctly folded structures [37].
2.6. Prediction of Secondary and Tertiary Structures
The secondary structure of the fusion proteins was predicted using the Garnier–Osguthorpe–Robson (GOR) IV method (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl ?page=npsa_gor4.html) [45]. Tertiary structures were generated with the I-TASSER server (https://zhanglab. ccmb.med.umich.edu/I-TASSER), and the reliability of each model was evaluated using the confidence score (C-score) provided by the server [37, 46]. The three-dimensional (3D) structure was analyzed using Molegro Molecular Viewer software.
2.7. Validation of the Tertiary Structure
The stereochemical integrity of the predicted 3D structures was assessed using Ramachandran plots generated by SWISS-MODEL (https://swissmodel.expasy. org/assess) [45]. Amino acid residues were classified as favored, allowed, or outlier to evaluate the reliability of the structural models.
2.8. Assessment of Antigenicity, Allergenicity, and Solubility
The antigenic potential of the chimeric protein was evaluated using VaxiJen v2.0 (http://www.ddg-pharmfac .net/vaxijen/VaxiJen/VaxiJen.html) [47]. Allergenicity was predicted using AlgPred (http://www.imtech.res.in/rag hava/algpred/), a tool that combines multiple prediction methods [48]. Additionally, the solubility of the protein upon expression in Escherichia coli (E. coli) was estimated using SOLpro (http://scratch.proteomics.ics.uci.edu/) [48].
2.9. Prediction of Physicochemical Properties
Key physicochemical properties of the protein, including molecular weight (MW), theoretical isoelectric point (pI), counts of positively and negatively charged residues, estimated half-life, extinction coefficient, aliphatic index, instability index, and grand average of hydropathicity (GRAVY), were computed using the ProtParam tool (http://web.expasy.org/protparam/) [45].
2.10. Optimization of the Chimeric Gene
To improve expression in E. coli, the coding sequence of the chimeric protein was reverse-translated and subjected to codon optimization using the European Bioinformatics Institute (EBI) tool (https://www.ebi.ac.uk /Tools/st/emboss_backtranseq/) [37, 49].
2.11. mRNA Structure Prediction
The mRNA secondary structure was predicted using the mfold web server (http://unafold.rna.albany.edu/?q= mfold) [37]. This analysis estimated the minimum free energy (MFE) and identified key structural elements, including hairpins and loops, which can influence mRNA stability and translation efficiency.
3. RESULTS
3.1. Gene Information
This in silico study was conducted between September and December 2025. Complete sequences for the SAG1 (336 aa, UniProt: C7E5T3), GRA1 (190 aa, UniProt: B9PHR1), and MIC4 (580 aa, UniProt: D8L550) antigens were retrieved from the UniProt database. All sequences correspond to the T. gondii RH strain. These antigens were selected based on their functional relevance and sequence length.
3.2. Transmembrane Domain and Signal Peptide Prediction
TMHMM analysis indicated that MIC4 and SAG1 lack transmembrane domains, whereas GRA1 contains a predicted transmembrane region. To ensure surface accessibility of epitopes and facilitate soluble recombinant expression, the transmembrane domain of GRA1 was excluded from the chimeric construct. Signal peptide prediction using SignalP 4.1 identified N-terminal signal peptides spanning residues 1–25 in MIC4 and SAG1 and residues 1–24 in GRA1. These signal peptide regions were not included in the selected epitope-rich fragments used for chimeric protein design. The predicted cleavage sites and D-scores for each antigen are provided in Supplementary Files 1 and 2 .
3.3. Prediction of B-cell and T-cell Epitopes
The epitope selection process was carried out in two main stages. Initially, the three selected antigens (SAG1, GRA1, and MIC4) were analyzed to predict potential B-cell epitopes, and the identified epitope sites are summarized in Table 1 . Next, each of these B-cell epitope sites was evaluated for the presence of T-cell epitopes. Fragments of 120 amino acids containing the highest number of T-cell epitopes with binding scores above 9 were prioritized as immunologically relevant regions, as summarized in Table 2 . Analysis of T-cell epitope prediction revealed multiple high-affinity MHC class I and class II binding peptides within these selected fragments. These regions contained the highest density of predicted MHC-binding epitopes across the evaluated alleles specific to the BALB/c mouse model. Given the critical role of Th1-mediated immune responses in controlling intracellular parasites such as T. gondii , these fragments were prioritized for chimeric vaccine construction. In parallel, the selected fragments exhibited favorable immunogenic properties, including high surface accessibility, antigenicity, hydrophilicity, and flexibility, representing overlapping B-cell and T-cell epitope-rich regions. To provide a clear overview, the overall workflow for B-cell and T-cell epitope prediction and selection is summarized in Figs. ( 1 and 2 ).
| B-cell Parameters | SAG1 | GRA1 | MIC4 |
|---|---|---|---|
| Bepipred Linear Epitope | 20-80, 80-145, 145-245, 245-300, 300-330 | 67-78, 87-98, 122-145, 170-180 | 70-95, 172-185, 208 222, 240-260, 305-320, 330-405, 417-445 |
| Beta-Turn | 45-65, 90-115, 125-140, 160-170, 180-265, 285-295, 305-315 | 65-80, 86-98, 108-112, 120-125, 135-145, 173-186 | 60-90, 105-133, 150-165, 200-220, 305-320, 330-345, 395-405, 420-445 |
| Accessibility | 60-100, 120-155, 165-210, 220-295 | 65-75, 87-90, 105-114, 119-125, 138-147, 167-180 | 175-200, 218-235, 270-278, 285-318, 360-381 |
| Flexibility | 60-115, 120-170, 190-290, 305-315 | 70-80, 85-95, 105-115, 120-128, 137-145, 165-182 | 70-97, 175-185, 205-245, 255-278, 305-318, 360-380, 400-410, 420-445 |
| Antigenicity | 25-120, 140-220, 280-300, 310-336 | 78-86, 90-103, 115-120, 132-137, 145-170 | 100-130, 150-170, 235-270, 280-305, 330-345, 440-453 |
| Hydrophilicity | 55-115, 125-140, 160-240, 250-320 | 67-75, 85-100, 106-113, 120-145, 155-180 | 70-95, 173-185, 207-222, 240-260, 305-320, 330-345, 395-407, 415-445 |
| Protein | Amino acid Start Position1 | Number of Binding Epitopes2 | Total3 | |||||
|---|---|---|---|---|---|---|---|---|
| Name | H2-Kd | H2-Ld | H2-Dd | I-Ad | I-Ed | MHC- I | MHC- II | |
| SAG1 | 242, 273 | -- | 190, 198, 206, 221, 227, 259, 261, 275, 288 | 181, 184, 195, 200, 217, 247, 260, 268, 278, 296, 300 | 181, 214, 221, 241, 267, 271, 279, 292, 298 | 11 | 20 | 31 |
| GRA1 | -- | -- | 71, 74, 122, 137 | 65, 81, 94, 114, 142, 144, 148, 151, 155 | 155, 165, 171 | 4 | 12 | 16 |
| MIC4 | 361, 378 | -- | 374, 378, 383, 417, 424, 457, 466, 470, 471 | 369, 398, 403, 432, 441, 445, 458, 462 | 363, 369, 379, 420, 428, 444, 459, 466, 469 | 11 | 17 | 28 |
Note: 1: Epitopes exhibiting a binding score greater than 9.
2: Number of high-scoring (score > 9) epitopes predicted to bind MHC-I and MHC-II within SAG1 (181-300), GRA1 (61-180), and MIC4 (361-480) antigens.
3: Total count of epitopes with scores above 9 in the SAG1 (181-300), GRA1 (61-180), and MIC4 (361-480) antigens.

Workflow for prediction and selection of B-cell and T-cell epitopes in SAG1, GRA1, and MIC4. Protein sequences were retrieved from public databases and analyzed for signal peptides and transmembrane helices. Linear B-cell epitopes were predicted based on antigenicity, flexibility, surface accessibility, hydrophilicity, and β-turn propensity. MHC class I and II T-cell epitopes were predicted based on binding affinity to reference human leukocyte antigen (HLA) alleles. High-scoring, overlapping epitopes were selected and assembled using appropriate linkers to generate the final chimeric SGM construct.
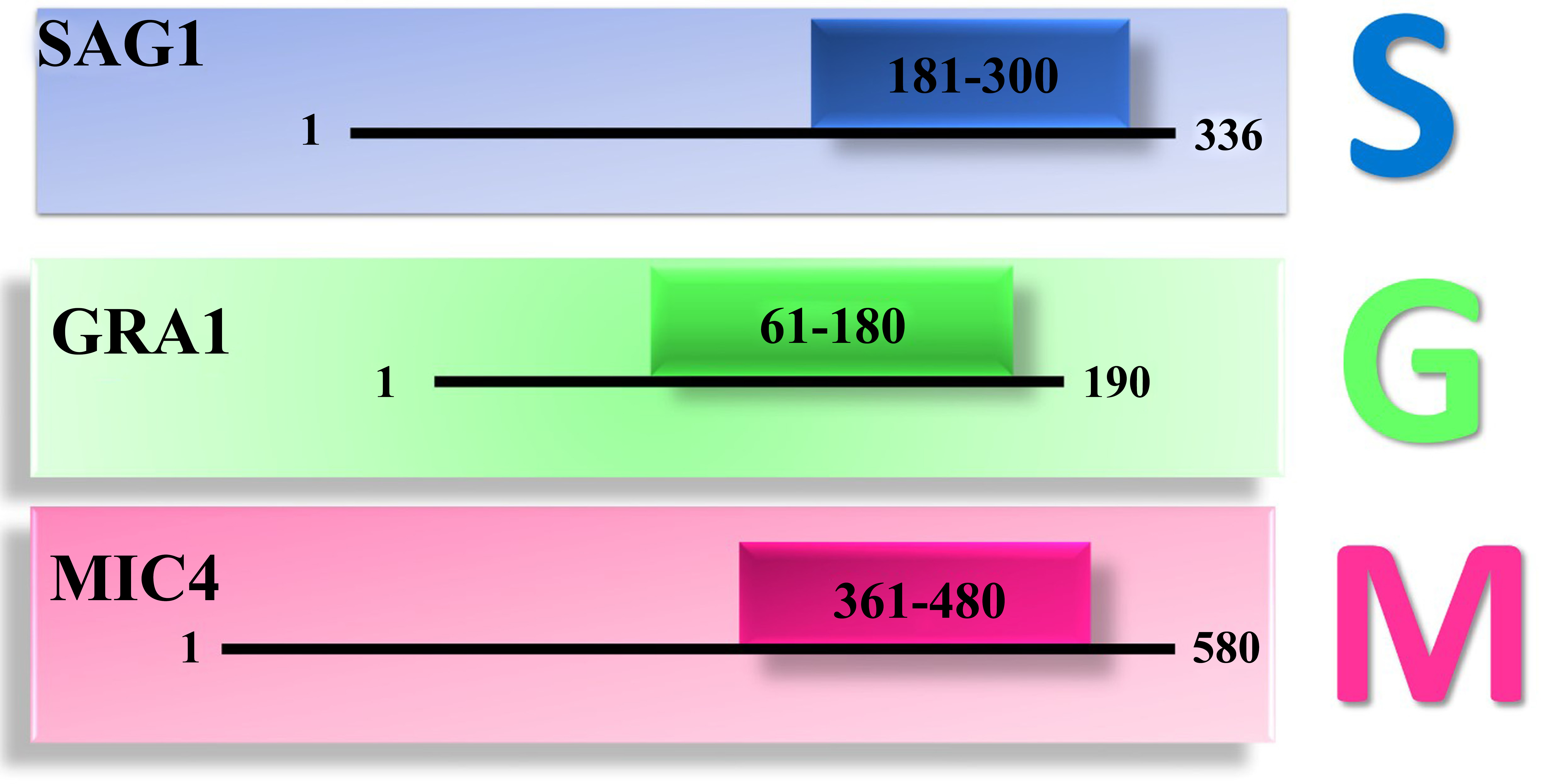
Identification of overlapping B- and T-cell epitope-rich regions in SAG1, GRA1, and MIC4 antigens. Linear B-cell epitopes and MHC class I and II T-cell epitopes were predicted for SAG1, GRA1, and MIC4 proteins using the Immune Epitope Database (IEDB) analysis resource. Regions containing overlapping B-cell epitopes and high-affinity MHC class I and II binding peptides were identified and mapped along the protein sequences. These epitope-dense regions were selected as immunodominant segments for inclusion in the chimeric SGM construct.
3.4. Segment Selection
To design the chimeric structure, fragments of 120 amino acids in length from the target antigens were selected: SAG1 (S181-E300), GRA1 (L61-D180), and MIC4 (F361-G480). Six chimeric arrangements (SMG, MSG, MGS, SGM, GMS, and GSM) were constructed from three antigenic fragments by a helix-forming linker containing the A(EAAAK)A motif. Among these candidates, the SGM construct (SAG1-GRA1-MIC4) showed the highest antigenicity and C-score.
3.5. Prediction and Analysis of Secondary and Tertiary Structures
The SGM chimeric protein, consisting of 395 amino acids, displayed a secondary structure comprising 18.23% extended strands, 46.56% random coils, and 35.19% α-helices (Fig. 3A and B). Tertiary structure modeling yielded a 3D model (Fig. 4A and B) with a C-score of -1.74, indicating moderate confidence in the predicted conformation.
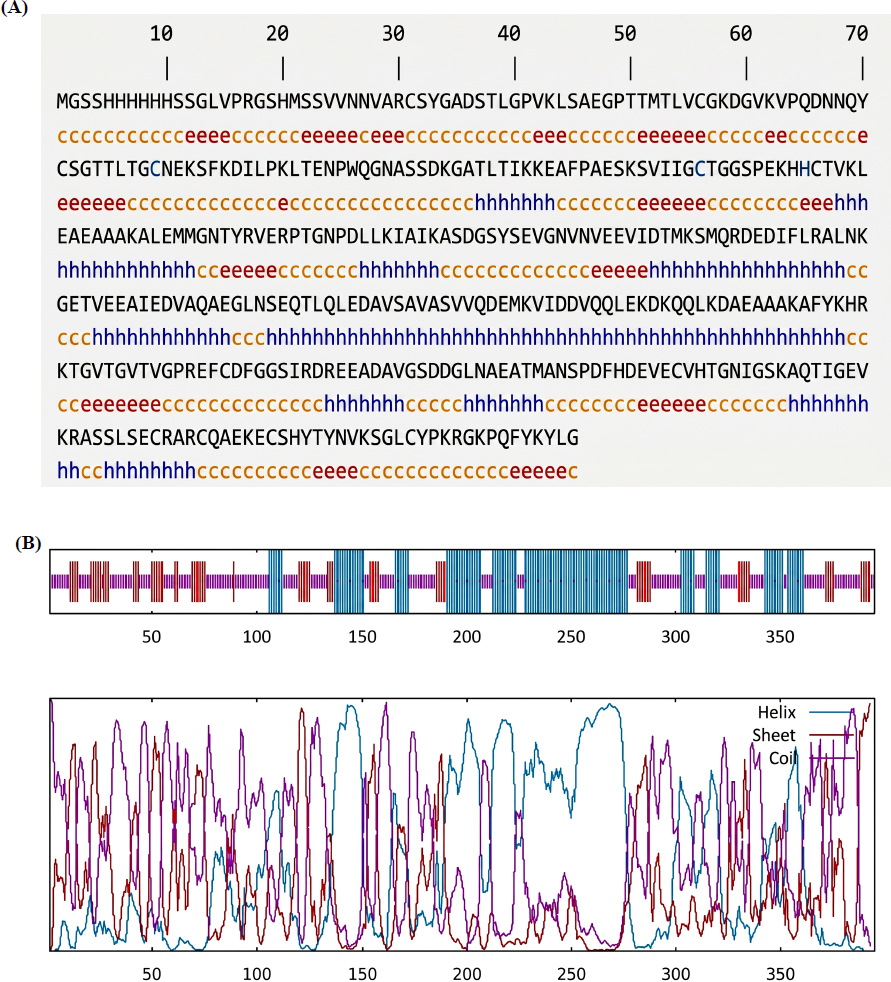
Secondary structure prediction of the SGM protein performed using the GOR IV online tool, classifying residues as α-helix (h), extended strand (e), or random coil (c). (B) Graphical representation of the predicted secondary structure distribution along the SGM protein sequence, illustrating the proportion and spatial arrangement of helices, strands, and coils, as generated by the GOR IV algorithm.
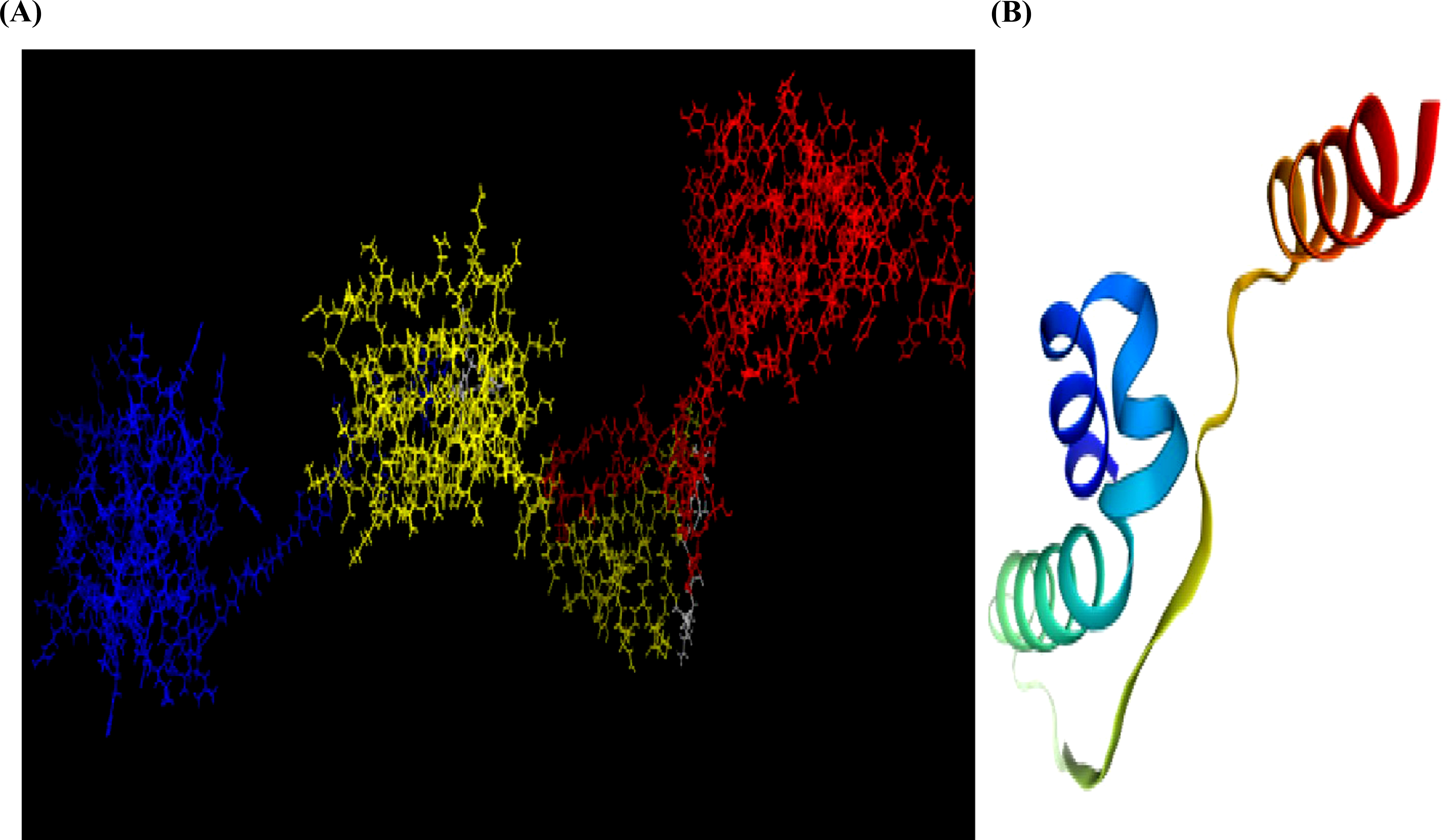
Predicted tertiary structure of the SGM protein generated using the I-TASSER server. SAG1, GRA1, and MIC4 segments are shown in red, yellow, and blue, respectively, while linker regions are shown in white. (B) Three-dimensional structural model of the SGM protein obtained using the SWISS-MODEL server, based on homology modeling. The model represents the overall folding and spatial arrangement of the chimeric construct.
3.6. Validation of the Tertiary Structure
Ramachandran plot analysis showed that 96.08% of residues were in favored regions, 3.92% in allowed regions, and 0% were outliers (Fig. 5).
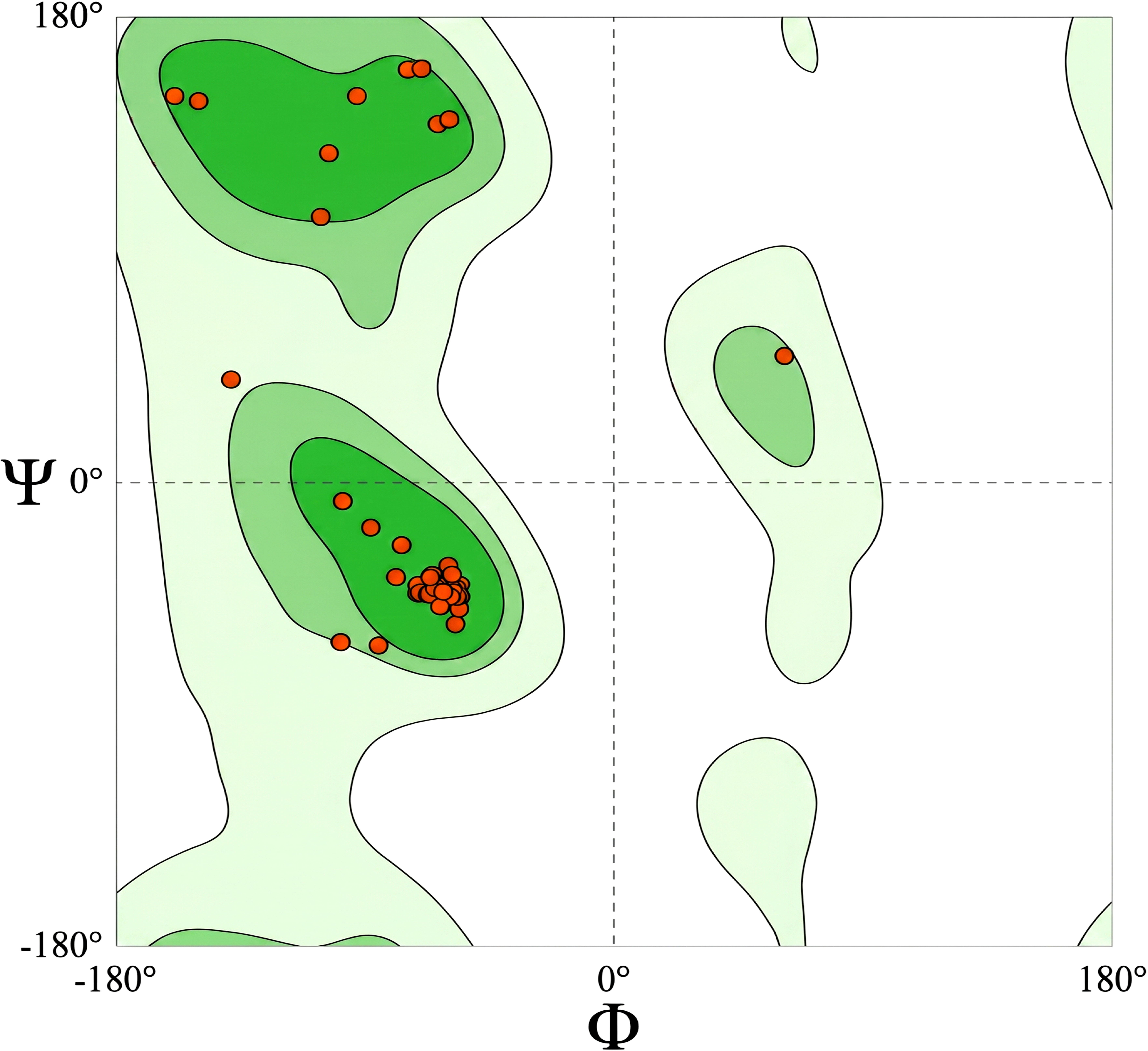
Validation of the SGM protein tertiary structure using a Ramachandran plot.
Ramachandran plot generated for the SWISS-MODEL-derived SGM protein structure to assess stereochemical quality. A total of 96.08% of amino acid residues were located in favored regions, while 3.92% were found in allowed regions, and no residues were detected in disallowed regions, indicating a reliable and structurally stable model.
3.7. Antigenicity, Allergenicity, and Solubility Evaluation
Antigenicity scores for six chimeric arrangements were calculated: SMG (0.6345), MSG (0.6218), MGS (0.6165), SGM (0.6394), GMS (0.6215), and GSM (0.6317). Based on antigenicity and structural analyses, the SGM was selected as the final construct. SGM was predicted to be non-allergenic, and its solubility score was 0.866.
3.8. Physicochemical Property Prediction
SGM exhibited a MW of 42.62 kDa and a pI of 5.53. The protein contained 58 negatively charged residues (Asp + Glu) and 45 positively charged residues (Arg + Lys). The predicted half-life was approximately 30 hours in mammalian reticulocytes, more than 20 hours in yeast, and more than 10 hours in E. coli. The instability index of 32.05 classified SGM as stable. The GRAVY score was –0.570, and the aliphatic index was 67.16, indicating good hydrophilicity and thermal stability.
3.9. Codon Optimization
The chimeric gene was reverse-translated and codon-optimized using the EBI server to enhance expression efficiency in E. coli by adapting codon usage to the host organism.
3.10. mRNA Secondary Structure Prediction
The best-predicted structure had a ΔG of –355.20 kcal/mol, and the first 10 nucleotides at the 5′ end did not form any stable hairpins or pseudoknots, indicating a minimal likelihood of secondary structure formation in this region (Fig. 6).
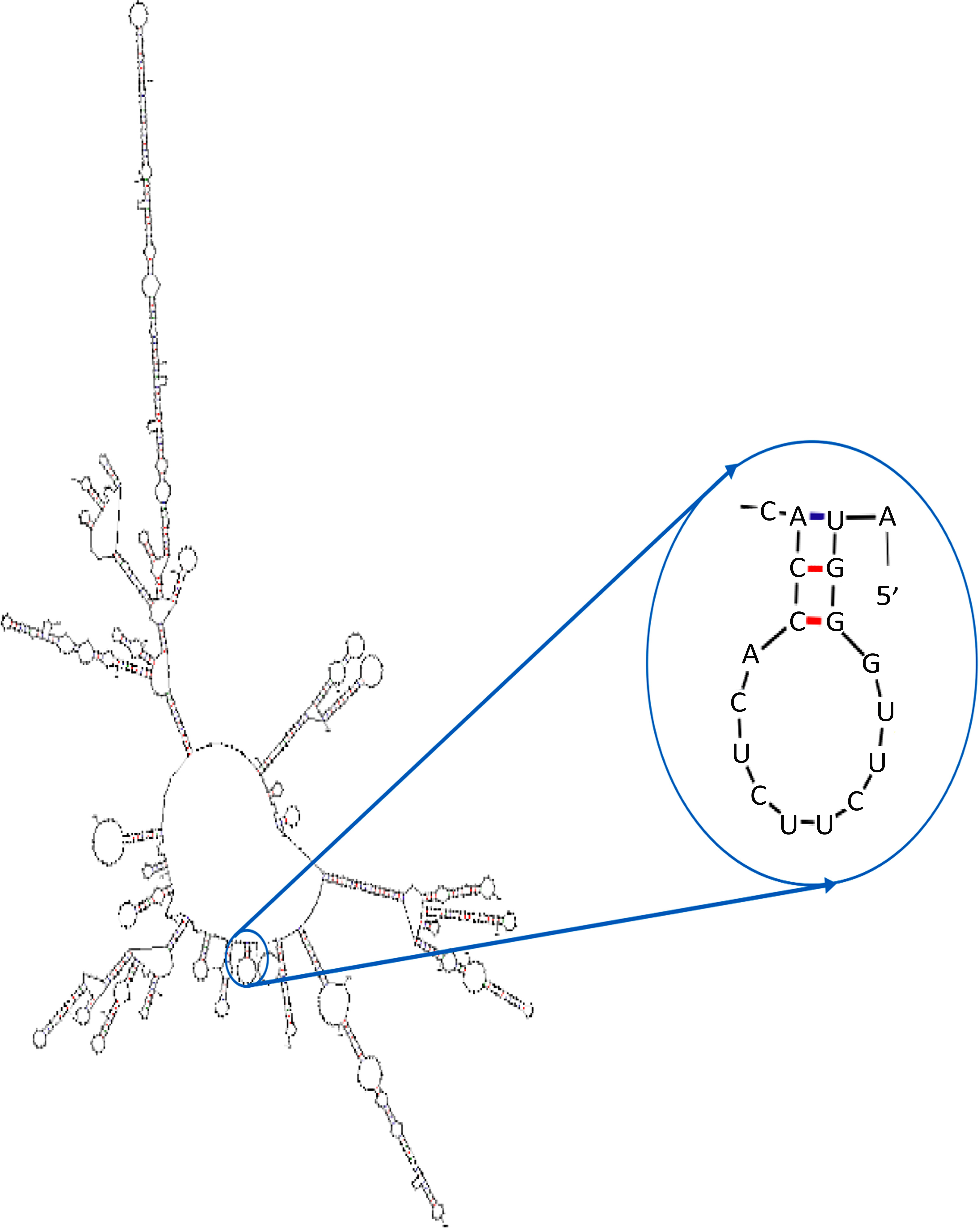
Predicted secondary structure of the SGM chimeric mRNA generated by the mFold server. The analysis demonstrated the absence of stable hairpin structures or pseudoknots at the 5′ end of the mRNA, suggesting favorable translational efficiency and structural stability of the mRNA construct.
4. DISCUSSION
In this study, a chimeric protein against T. gondii was designed by combining the dominant immunogenic regions of SAG1, GRA1, and MIC4 using an immunoinformatics approach. Although any antigen could be considered as a candidate using in silico methods, the selection of SAG1, GRA1, and MIC4 was driven by predefined biological and immunological criteria rather than antigen availability. In particular, these points were considered: (1) antigenic diversity by combining surface (SAG1) and secretory (GRA1 and MIC4) proteins; (2) prioritizing multi-antigen vaccine design over single-antigen approaches for broader immune coverage and more robust protection against toxoplasmosis [50-52]; and (3) selecting antigens expressed in all three stages of the parasite's life cycle to ensure a comprehensive immune response, as stage-specific antigens usually provide protection limited to that stage [53]. SAG1 is a major surface antigen for tachyzoites [54], while MIC4 and GRA1 are expressed in all three life stages of the parasite (i.e., the sporozoites, bradyzoites, and tachyzoites) [31, 32]. In addition, the selection was supported by their well-documented roles in parasite infectivity and immune stimulation. SAG1 stimulates both humoral and cellular immunity [37, 55]; GRA1 is involved in the formation of parasitophorous vacuoles and parasite survival [31, 56]; and MIC4 contributes to parasite movement, recognition, and attachment to the host cell, a critical step in invasion [32]. Collectively, these antigens support the induction of coordinated Th1/CD8+ T cell–mediated and humoral immune responses, which are essential for controlling acute infection and limiting parasite persistence during chronic toxoplasmosis [32, 57–60]. Recent experimental studies have demonstrated that the selected antigens (SAG1, GRA1, and MIC4) play important roles in host–parasite interaction and exhibit confirmed immunogenic and protective potential in vivo. For instance, vaccination with virus-like particles containing the SAG1 antigen demonstrated robust humoral immune responses, including significant elevations in IgG, IgG1, IgG2a, and IgA, along with increased production of Th1-associated cytokines such as IFN-γ and TNF-α. Importantly, this strategy resulted in up to 75% protection against challenge infection in a murine model, highlighting the strong protective capacity of SAG1-based formulations [61]. Similarly, a multicomponent DNA vaccine encoding both SAG1 and GRA1 induced significantly enhanced serum IgG responses and elevated Th1 cytokines (IFN-γ and IL-2), leading to prolonged survival in BALB/c mice following T. gondii challenge [62]. These findings emphasize the synergistic immunological effect of combining SAG and GRA antigens within a single vaccine platform. More recently, a multivalent DNA vaccine incorporating SAG1, SAG3, MIC4, GRA5, GRA7, AMA1, and BAG1 demonstrated significantly enhanced humoral and cellular immune responses compared with controls, further supporting the contribution of MIC family proteins-particularly MIC4-to broadening protective immunity [63]. In addition, recombinant multi-epitope protein constructs incorporating SAG1 and GRA1 (together with other antigens) have been experimentally expressed and immunologically evaluated in mice, resulting in strong antigen-specific antibody production and dominant Th1-biased cellular responses [51]. Notably, these experimental multi-epitope approaches parallel the conceptual framework of the present in silico design, further supporting the translational feasibility of epitope-based chimeric constructs derived from these antigens. Collectively, these studies provide compelling experimental validation for selecting SAG1, GRA1, and MIC4 as rational immunogenic targets. They demonstrate that these antigens are capable of inducing strong humoral and Th1-oriented cellular immunity and conferring measurable protection in murine models. Therefore, the overlapping B- and T-cell epitopes identified in our computational analysis are biologically plausible and supported by prior in vivo evidence, warranting further experimental evaluation of the proposed chimeric vaccine candidate.
Previous studies have evaluated T. gondii vaccines using full-length antigens such as SAG1, GRA1, MIC4, or combinations thereof, in various experimental settings, demonstrating partial protection and induction of humoral and cellular responses [31, 32, 50, 64, 65]. The novelty of the present study lies in the rational epitope-based design strategy, rather than the use of full-length antigens. This in silico strategy allows precise selection of immunodominant regions and facilitates subsequent structural and immunological evaluation prior to experimental validation. Collectively, our design complements previous experimental efforts and provides a framework for constructing chimeric vaccines with potentially improved efficacy and safety.
This study identified 120-amino-acid fragments of SAG1, GRA1, and MIC4 that are rich in both B-cell and T-cell epitopes. These fragments exhibit overlapping B- and T-cell epitope regions, allowing a single fragment to induce both humoral and cellular immune responses, potentially enhancing protection against T. gondii. The high density of predicted MHC class I and II binding peptides supports robust Th1-mediated cellular immunity, while favorable properties such as surface accessibility, antigenicity, hydrophilicity, and flexibility underscore their suitability for a chimeric vaccine. Overlapping epitopes were identified and linked using a rigid linker with the sequence A(EAAAK)nA to connect the SAG1, GRA1, and MIC4 domains. Rigid linkers are generally more effective than flexible linkers in maintaining structural independence and proper folding of individual domains, minimizing potential steric hindrance or epitope masking [66, 67]. The number of EAAAK repeats (n) was optimized through structural modeling to provide sufficient spatial separation between adjacent epitopes while preserving overall stability and compactness of the chimeric construct. Using the three antigens linked by A(EAAAK)nA, six possible conformations were generated. The final structure (SGM) was selected based on multiple criteria, including antigenicity and secondary and tertiary structure quality. While high antigenicity ensures recognition by the immune system, structural integrity is equally important for protein stability and proper function. Tertiary structures were modeled using I-TASSER, which reports five models with confidence quantified by a C-score (ranging from –5 to 2, with higher values indicating greater confidence). SGM exhibited the highest C-score (–1.74), indicating it as the most reliable structure for further investigation. Evaluation of the tertiary structure showed that 96.08% of residues are located in favorable regions of the Ramachandran plot, demonstrating excellent stereochemical quality and appropriate folding. The SGM chimeric protein, consisting of 395 amino acids, displayed a secondary structure composed of 35.19% α-helices, 18.23% extended strands, and 46.56% random coils. This protein exhibits a relatively high proportion of random coils, which, rather than being detrimental, may be beneficial for vaccine design. Random coil regions are generally flexible and surface-exposed, facilitating accessibility of B-cell epitopes and enhancing antibody recognition. Part of this random coil content also corresponds to linker sequences [A(EAAAK)nA], which maintain independent folding of the antigenic domains. At the same time, the presence of α-helices and extended strands contributes to protein stability and enhanced interaction with antibodies [68], supporting its suitability as a chimeric vaccine candidate.
The predicted solubility of 0.866 for this protein was made assuming expression in E. coli, as this host is widely used for recombinant antigen production. However, solubility may vary in alternative expression systems. In general, proteins with high solubility are easier to purify and are less likely to form inclusion bodies. The predicted MW of the protein (42.62 kDa) is within the range generally considered suitable for expression and purification in bacterial systems. For example, recombinant SAG1 constructs used in vaccine and diagnostic studies typically range from ~30 kDa to ~48 kDa, depending on the inclusion of fusion tags, while recombinant GRA1 is usually smaller, around ~22 kDa. These observations indicate that the molecular size of SGM is suitable for efficient expression, purification, and downstream formulation in bacterial systems. Knowledge of the protein’s MW and pI can also inform buffer selection and solubility optimization, which are critical for maintaining protein stability and immunogenicity in vaccine formulations. An instability index value of less than 40 indicates that the SGM has adequate stability. The GRAVY value of -0.570 indicates its highly hydrophilic nature, which increases solubility. Hydrophilic proteins tend to expose antigenic regions more readily, improving recognition by B cells and antibody accessibility. The high antigenicity score predicted by VaxiJen indicates strong potential to stimulate immune responses, while allergenicity assessment revealed no predicted allergenic motifs, suggesting a low risk of IgE-mediated adverse reactions. Although antigenicity and allergenicity were predicted using established in silico tools, such predictions are inherently limited and cannot fully reflect in vivo immune responses. Therefore, these results should be considered as preliminary indicators to guide construct selection, and experimental validation will be essential to confirm immunogenicity and safety. Collectively, these physicochemical properties indicate that SGM is not only structurally stable and soluble but formulation-friendly, supporting its suitability for vaccine delivery and immunogenic efficacy in vivo.
Codon optimization, together with mRNA secondary structure analysis, was employed to improve the translational efficiency of the construct. The optimized mRNA exhibited a low minimum free energy at the 5′ end, reducing the likelihood of inhibitory hairpin formation and facilitating efficient protein expression. These results confirm the suitability of SGM design using bioinformatics tools.
5. LIMITATIONS AND FUTURE PERSPECTIVES
Despite the promising immunological and structural characteristics predicted for the SGM chimeric vaccine candidate, several limitations of the present study should be acknowledged. First, this study is entirely based on in silico analyses. Although immunoinformatics approaches are powerful tools for preliminary vaccine design and help reduce time and cost, computational predictions cannot fully capture the complexity of immune responses in vivo. Therefore, experimental validation through in vitro assays and animal models is essential to confirm the immunogenicity, safety, and protective efficacy of the proposed vaccine candidate. Second, T-cell epitope prediction was primarily performed using MHC class I and II alleles specific to the BALB/c mouse model. While this choice is appropriate for preclinical evaluation, it may limit the direct extrapolation of the findings to genetically diverse human populations. Future studies should extend epitope prediction and population coverage analyses to a broader range of human HLA alleles. Third, the present study focused mainly on linear B-cell epitopes and predicted T-cell epitopes. Conformational B-cell epitopes, which may play a significant role in antibody-mediated immunity, were not explicitly evaluated. Incorporation of conformational epitope prediction and experimental antibody-binding assays would further strengthen the vaccine design. Another consideration is immune tolerance, which may occur if some epitopes are poorly immunogenic or resemble host sequences. This factor could influence the magnitude and quality of both humoral and cellular responses. Although the epitope selection strategy aimed to minimize these effects by prioritizing highly antigenic and accessible regions, experimental studies are required to assess their actual impact. Future in vivo validation of the SGM chimeric vaccine will be conducted in accordance with institutional ethical guidelines for animal experimentation and under appropriate biosafety conditions to ensure humane treatment and containment of T. gondii. Furthermore, the use of appropriate adjuvants can play an important role in optimizing and sustaining immune responses. This chimeric construct was designed without an intrinsic adjuvant sequence. This decision was made to focus on epitope selection and structural optimization of the antigen itself and to avoid potential interference of adjuvant domains with protein folding or epitope accessibility. In practical vaccine development, adjuvants are typically introduced during formulation rather than genetic fusion, allowing flexibility in modulating immune responses. Future experimental studies will explore appropriate adjuvant systems to enhance the immunogenicity of the SGM construct.
CONCLUSION
This study presents the in silico design of a chimeric vaccine candidate containing immunogenic epitopes from SAG1, GRA1, and MIC4 antigens. Structural and physicochemical analyses suggest that the SGM chimeric protein is potentially stable, antigenic, and capable of eliciting both humoral and cellular immune responses. These findings provide a rational basis for vaccine design against toxoplasmosis; however, experimental validation, including in vitroand in vivostudies, is essential to confirm immunogenicity and protective efficacy.
AUTHORS' CONTRIBUTIONS
The authors confirm contribution to the paper as follows: T.N: Conceived the study and designed the study protocol; T.N: The supervisor of this research; E.N and M.F: Performed the bioinformatics analysis; T. N: Drafted the manuscript; T.N: Critically revised the manuscript; and all authors read and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| T. gondii | = Toxoplasma gondii |
| IEDB | = Immune Epitope Database |
| MW | = Molecular Weight |
| pI | = Isoelectric Point |
| SAGs | = Surface Antigens |
| GRAs | = Dense Granule Proteins |
| ROPs | = Rhoptry Proteins |
| MICs | = Microneme Proteins |
| MHC | = Major Histocompatibility Complex |
| UniProt | = Universal Protein Resource |
| GOR | = Garnier–Osguthorpe–Robson |
| 3D | = Three Dimensional |
| C-score | = Confidence score |
| GRAVY | = Grand Average of Hydropathicity |
| EBI | = European Bioinformatics Institute |
| MFE | = Minimum Free Energy |
| HLA | = Human Leukocyte Antigen |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethical Committee of Dezful University of Medical Sciences (The code of ethics of this plan is IR.DUMS.REC.1404.060).
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
ACKNOWLEDGEMENTS
The authors would like to thank the Infectious and Tropical Diseases Research Center, Dezful University of Medical Sciences, Dezful, Iran, for their support, cooperation, and assistance throughout the period of the study (IN&TR-404060-1404).

