All published articles of this journal are available on ScienceDirect.

In silico Analysis of Toxoplasma gondii Surface and Secretory Proteins for the Design of a Novel Chimeric Vaccine
Authors Info & Affiliations
Abstract
Background
This study focuses on the Toxoplasma gondii (T. gondii) antigens ROP18, SAG1, and MIC13, which play key roles in pathogenesis, immune evasion, and host invasion. The aim was to design a novel chimeric antigen combining these proteins as a potential vaccine candidate against T. gondii.
Methods
Fragments of ROP18 (Q101–E300), SAG1 (P61–G160), and MIC13 (D171–R320) were linked using a rigid A(EAAAK)A linker. Bioinformatics analyses predicted various properties of the chimeric protein RSM1, including transmembrane domains, B- and T-cell epitopes, secondary and tertiary structures, antigenicity, physicochemical traits, codon optimization, and mRNA structure.
Results
RSM1 consists of 485 amino acids and has an antigenicity score of 0.6694. The aliphatic index, instability index, and GRAVY score were 68.66, 54.19, and –0.639, respectively. Structural predictions supported RSM1’s potential as a vaccine candidate. The most stable tertiary structure had a ΔG of –524.80 kcal/mol, with no stable hairpins or pseudoknots at the mRNA 5′ end, suggesting favorable translation.
Discussion
The bioinformatics analyses indicate that RSM1 possesses favorable antigenic and structural properties, supporting its potential as a multi-epitope vaccine candidate. Its predicted stability and translation efficiency suggest practical viability, although the moderate instability index points to the need for further optimization.
Conclusion
RSM1 represents a promising in silico-designed vaccine candidate against T. gondii. This study lays the groundwork for subsequent experimental evaluations to determine its immunogenicity and protective efficacy in vivo.
1. INTRODUCTION
Toxoplasma gondii (T. gondii) is an obligate intracellular zoonotic parasite reported in a variety of warm-blooded and even cold-blooded animals [1-7]. This protozoan is recognized as a significant food-borne parasite capable of infecting humans through the consumption of contaminated drinking water or raw/unwashed vegetables harboring mature oocysts, as well as through the ingestion of undercooked or raw meat harboring tissue cysts [8, 9].
The parasite’s life cycle comprises three stages: tachyzoite, bradyzoite, and sporozoite [10]. Tachyzoites are responsible for the acute phase, while bradyzoites cause the chronic phase of the disease [11]. Toxoplasmosis usually remains asymptomatic in immunocompetent individuals. However, in immunocompromised patients, chronic infection can reactivate, potentially resulting in severe conditions such as toxoplasmic encephalitis, which can be fatal [12]. In addition, pregnant women face risks associated with this infection, including miscarriages, congenital defects, and chorioretinitis [13, 14].
Despite recent significant advances in the biology of T. gondii, the treatment of toxoplasmosis is still limited to controlling the tachyzoite stage, and current drugs cannot eliminate the parasitic cyst [15]. Chemotherapeutic options for T. gondii infection are limited. The two major drugs that are used in the treatment of acute toxoplasmosis are pyrimethamine and sulfadiazine, which are not enough when they are used alone [15]. Additionally, these drugs have various side effects [16]. Therefore, the development of a peptide-based vaccine using epitopes that stimulate humoral and cellular immune responses against Toxoplasma is necessary to control the infection. For this purpose, parasite antigens with high immunogenicity are targeted [17]. Among T. gondii antigens, surface antigens (SAGs), rhoptry proteins (ROPs), microneme proteins (MICs), and dense granule proteins (GRAs) are considered potential antigenic targets [18]. Given the complexity of the life cycle, diversity, and variability of intracellular parasite antigens such as T. gondii, vaccination should stimulate the immune system against multiple antigens [19]. MIC13 protein is crucial for the parasite’s dissemination within the host [20]. Additionally, ROP18, a secretory antigen expressed exclusively during the tachyzoite stage, plays a vital role in parasite pathogenesis [21]. Meanwhile, SAG1 is a major surface antigen found exclusively in the tachyzoite phase, contributing to immune evasion and attenuation of virulence [22]. Consequently, this study focuses on designing a vaccine candidate against T. gondii by utilizing T-cell and B-cell epitopes derived from the antigens ROP18, SAG1, and MIC13 using in silico approaches.
2. MATERIALS AND METHODS
2.1. Sequence Retrieval
In 2024, amino acid sequences for ROP18, SAG1, and MIC13 from the T. gondii RH strain were sourced from the Universal Protein Resource (UniProt) (http://www. uniprot.org/) in FASTA format. Among the available sequences, the longest sequence was selected for each antigen (ROP18 [UniProt: Q2PAY2], SAG1 [UniProt: C7E5T3], and MIC13 [UniProt: H9BC62]). These sequences were utilized for subsequent computational analyses.
2.2. Prediction of Transmembrane Domains and the Signal Peptides
The transmembrane topology of the selected proteins was assessed using the TMHMM server, which employs a Hidden Markov Model approach [23]. Both the UniProt and SignalP servers were used to evaluate the presence of signal peptides within the proteins [24].
2.3. B-cell Epitope Prediction
B-cell epitopes were predicted using the Immune Epitope Database (IEDB). This server considers several parameters to increase prediction accuracy, including linear epitope prediction [25], β-turn prediction [26], flexibility [27], antigenicity [28], hydrophilicity [29], and surface accessibility [30].
2.4. MHC-I and MHC-II Epitopes
To identify potential epitopes that bind to major histocompatibility complex (MHC) molecules, antigen sequences were analyzed using the IEDB server, with a focus on the BALB/c mouse strain alleles. Following IEDB guidelines, peptides of 9 amino acids in length were preferred, and epitopes were ranked based on their predictive scores, ranging from 0 to 10 [31].
2.5. Fusion Peptide Design
For the design of a chimeric protein antigen, immunogenic epitopes derived from ROP18, SAG1, and MIC13 were strategically linked via a helical structure containing the A(EAAAK)A motif, which promotes an optimal spatial arrangement of the epitopes [32]. The A(EAAAK)A motif is an empirical rigid linker that maintains proper spacing between domains [24].
2.6. Prediction of Secondary and Tertiary Structures
The Garnier-Osguthorpe-Robson (GOR) IV (https://npsa- prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page= npsa_gor4.html) method was employed to predict the secondary structures, assessing the probabilities of α-helices, β-sheets, and random coils [33]. The ITASSER server (https://zhanglab.ccmb.med.umich.edu/I-TASSER) was utilized to generate three-dimensional (3D) conformations of the sequences, resulting in confidence scores known as confidence scores (C-scores) [24, 34]. This server predicts protein structure and function. The C-score, one of the most important outputs of the server, estimates the accuracy and quality of the predicted models [34]. Additional three-dimensional (3D) models of the protein sequences were produced using the Molegro Molecular Viewer software, facilitated through SWISS-MODEL [35].
2.7. Surveying Validation of the Tertiary Structure
The validity of the predicted 3D structure of the RSM1 protein was confirmed using the Ramachandran plot generator included in the SWISS-MODEL program to assess stereochemical quality (https://swissmodel.expasy. org/assess) [33]. This tool visualizes energetically favored regions of backbone dihedral angles relative to amino acid residues in a protein structure [33].
2.8. Antigenicity, Allergenicity, and Solubility Evaluation
The antigenicity of various antigen fragment configurations was analyzed using VaxiJen v.2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), where a score above 0.5 indicates potential antigenic properties, reflecting a server accuracy of between 70% and 89% depending on the target organism [36]. Allergenicity was evaluated through the Algpred server (http://www.imtech.res.in/raghava/algpred/), which offers predictions with approximately 85% accuracy at a threshold of –0.4 [37]. Additionally, protein solubility propensity was estimated using the SOLpro web server (http://scratch.proteomics.ics.uci.edu/), particularly for expression in Escherichia coli (E. coli) [37].
2.9. Prediction of Physical and Chemical Properties
Physicochemical properties of the chimeric protein were predicted using the ProtParam server (http://web.expasy. org/protparam/), which predicts a set of protein features including the molecular weight (MW), isoelectric point, total number of positively and negatively charged residues, estimated half-life, extinction coefficient, aliphatic index, instability index, grand average of hydropathicity (GRAVY), and other physical and chemical properties [33].
2.10. Optimization of the Chimeric Gene
To enhance protein expression efficiency, codon optimization techniques were applied in this study, utilizing the European Bioinformatics Institute (EBI) database (https://www.ebi.ac.uk/Tools/st/emboss_backtranseq/) for reverse translation and codon optimization [24, 38].
2.11. mRNA Structure Prediction
The mfold tool (http://unafold.rna.albany.edu/?q=mfold) was utilized to evaluate the free energy of the 5′ ends of the mRNA derived from the recombinant gene, providing insights into the thermodynamic stability of the mRNA molecule [24].
3. RESULTS
3.1. Information about Genes
The investigation identified the most complete sequences for the ROP18 (UniProt: Q2PAY2), SAG1 (UniProt: C7E5T3), and MIC13 (UniProt: H9BC62) antigens, selected based on their substantial lengths of 554, 336, and 468 amino acids, respectively.
3.2. Prediction of Transmembrane Domains and the Signal Peptides
Using the TMHMM server, none of the selected antigens (ROP18, SAG1, and MIC13) were found to contain transmembrane domains. This finding highlights the importance of exposed epitopes on cell surfaces for effective immunogenicity and informs the design of chimeric antigens to exclude transmembrane domains. The SignalP 4.1 server identified potential signal peptide regions at positions 1–47 for ROP18, 1–25 for SAG1, and 1–22 for MIC13.
3.3. B-cell and T-cell Epitope Prediction
Initially, T cell epitopes of ROP18, MIC13, and SAG1 antigens were predicted separately. To assess the affinity of these epitopes for MHC class I (H2-Kd, H2-Ld, H2-Dd) and class II (I-Ad, I-Ed) alleles specific to the BALB/c mouse strain, the antigen sequences were submitted to the IEDB web server. The server predicts peptides of 9 amino acids in length, assigning scores ranging from 0 to 10. For each antigen, the fragment with the highest score was selected. Subsequently, B-cell epitopes were selected using IEDB data, considering factors such as accessibility, hydrophilicity, antigenicity, flexibility, linear epitope prediction, and beta-turn propensity. Considering that Th1-type responses are more critical than Th2 responses in infections with intracellular parasites such as Toxoplasma, a 170-amino-acid fragment of each antigen containing multiple strong B- and T-cell epitopes, with preference given to strong T-cell epitopes, was selected as the final epitope. Epitopes were chosen from the initial candidate pool based on the following criteria: highest binding affinity scores to MHC alleles (score > 9), overlap between B- and T-cell epitope-rich regions, surface accessibility, antigenicity, hydrophilicity, flexibility, and distribution across different protein domains to maximize immune coverage. Tables 1 and 2 present the predicted B- and T-cell epitopes identified through analyses using various servers.
3.4. Segment Selection
Segments of the ROP18 (Q101-E300), SAG1 (P61-G160), and MIC13 (D171-R320) proteins were strategically selected for the construction of a chimeric antigen. The lengths of these segments were 200, 100, and 150 amino acids, respectively.
| B-cell Parameters | ROP18 | SAG1 | MIC13 |
|---|---|---|---|
| Bepipred Linear Epitope | 20-110, 125-140, 180-220, 240-340, 355-410, 515-550 | 20-80, 80-145, 145-245, 245-300, 300-330 | 70-95, 172-185, 208-222, 240-260, 305-320, 330-405, 417-445 |
| Beta-Turn | 40-145, 165-265, 285-380, 400-520 | 45-65, 90-115, 125-140, 160-170, 180-265, 285-295, 305-315 | 60-90, 105-133, 150-165, 200-220, 305-320, 330-345, 395-405, 420-445 |
| Accessibility | 50-190, 220-320, 445-550 | 60-100, 120-155, 165-210, 220-295 | 175-200, 218-235, 270-278, 285-318, 360-381 |
| Flexibility | 40-340, 370-450, 480-550 | 60-115, 120-170, 190-290, 305-315 | 70-97, 175-185, 205-245, 255-278, 305-318, 360-380, 400-410, 420-445 |
| Antigenicity | 20-40, 60-155, 190-285, 300-420, 470-540 | 25-120, 140-220, 280-300, 310-336 | 100-130, 150-170, 235-270, 280-305, 330-345, 440-453 |
| Hydrophilicity | 40-145, 165-340, 360-460, 480-550 | 55-115, 125-140, 160-240, 250-320 | 70-95, 173-185, 207-222, 240-260, 305-320, 330-345, 395-407, 415-445 |
| Protein | Amino Acid Start Position1 | - | - | Number of Binding Epitopes2 | Total3 | |||
|---|---|---|---|---|---|---|---|---|
| Name | H2-Kd | H2-Ld | H2-Dd | I-Ad | I-Ed | MHC- I | MHC- II | |
| ROP18 | 115, 153, 204 | -- | 164, 172, 196, 213, 233, 250, 255, 259, 261, 287 | 101, 132, 142, 147, 153, 177, 201, 225, 227, 237, 239, 250, 251, 267, 277, 279, 292, 294 | 110, 115, 116, 120, 122, 123,145, 151, 154, 156, 160, 161, 162, 163, 166, 170, 173, 199, 230, 239, 242, 278 | 13 | 40 | 53 |
| SAG1 | 79 | -- | 93, 109,139,149 | 62, 66, 68, 77, 90, 96, 109, 112, 119, 138, 140, 156 | 73, 78, 96, 110, 142, 157 | 5 | 18 | 23 |
| MIC13 | -- | -- | 190, 222, 228, 229, 308 | 182, 195, 200, 222, 227, 234, 237, 244, 248, 257, 265, 271, 284, 299 | 173, 183, 184, 185, 186, 193, 198, 297, 298, 300, 302, 303, 319 | 5 | 27 | 32 |
2: The number of epitopes with a score higher than 9 that bind to MHC-I and MHC-II in ROP18 (101-300), SAG1 (61-160), and MIC13 (171-320) antigens.
3: Total number of epitopes with a score higher than 9 in ROP18 (101-300), SAG1 (61-160), and MIC13 (171-320) antigens.
3.5. Prediction and Analysis of Secondary and Tertiary Structures
Analysis of the secondary structure of the composite protein RSM1, composed of 485 amino acids, revealed a secondary structure comprising 36.08% alpha-helix, 49.69% random coil, and 14.23% extended strand (Fig. 1A and B). The 3D structure model is shown in Fig. (2A and B), with a C-score of –2.00, indicating moderate model quality.
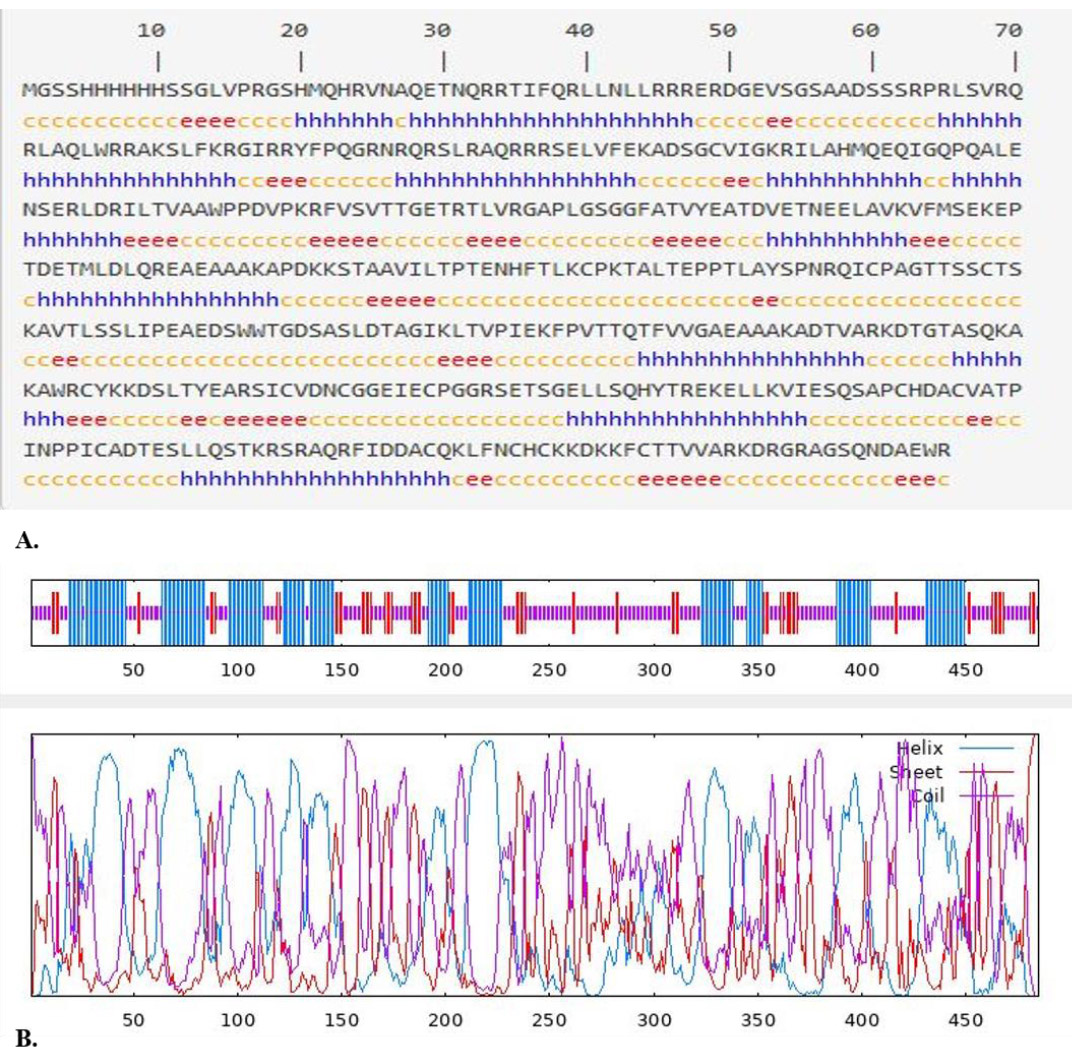
(A). Prediction of the secondary structure of RSM1 by GOR IV online service (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html). h=helix, e=extended strand, and c=coil, and (B) Graphical results for secondary structure prediction of RSM1 protein by GOR IV.
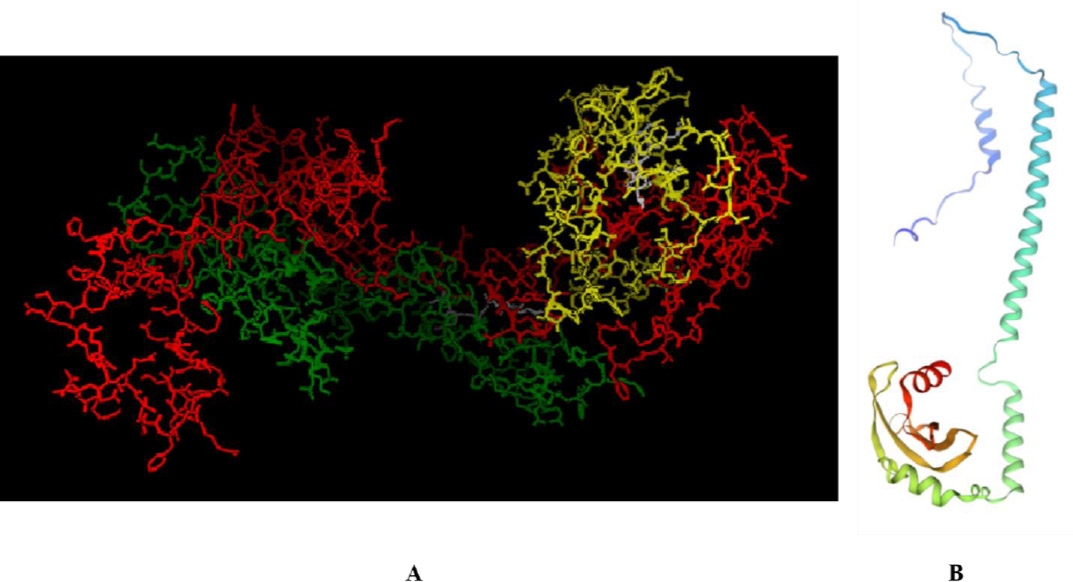
(A) Prediction of the tertiary structure of RSM1 using I-TASSER service (https://zhanglab.ccmb.med.umich.edu/I-TASSER) (ROP18: red, SAG1: yellow, MIC13: green, and linker: white) and (B) 3D model by the SWISS-MODEL server (https://swissmodel.expasy.org/assess).
3.6. Tertiary Structure Validation
The validation of the tertiary structure was performed using the Ramachandran plot, which showed that 91.46% of the residues were situated in the favored regions, 4.52% in allowed regions, and 4.02% were identified as outliers (Fig. 3).
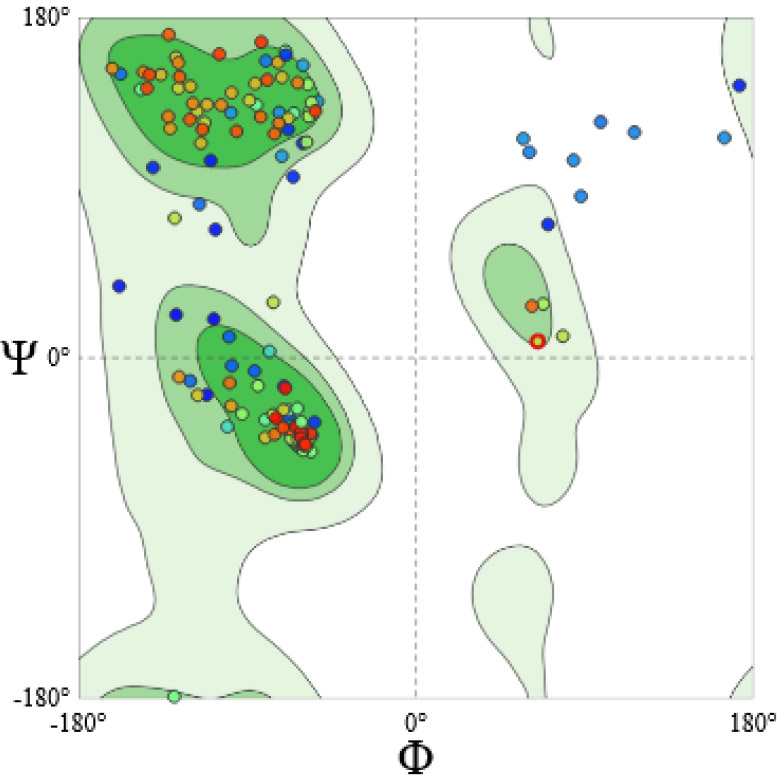
Validation of the tertiary structure of RSM1 protein using Ramachandran plot (https://swissmodel.expasy.org/assess). The analysis of Ramachandran plot statistics for the initial model revealed that 91.46% of amino acid residues from the structure modeled by SWISS-MODEL were incorporated in the favored regions; whereas only 4.52% and 4.02% are in allowed and outlier regions of the plot, respectively.
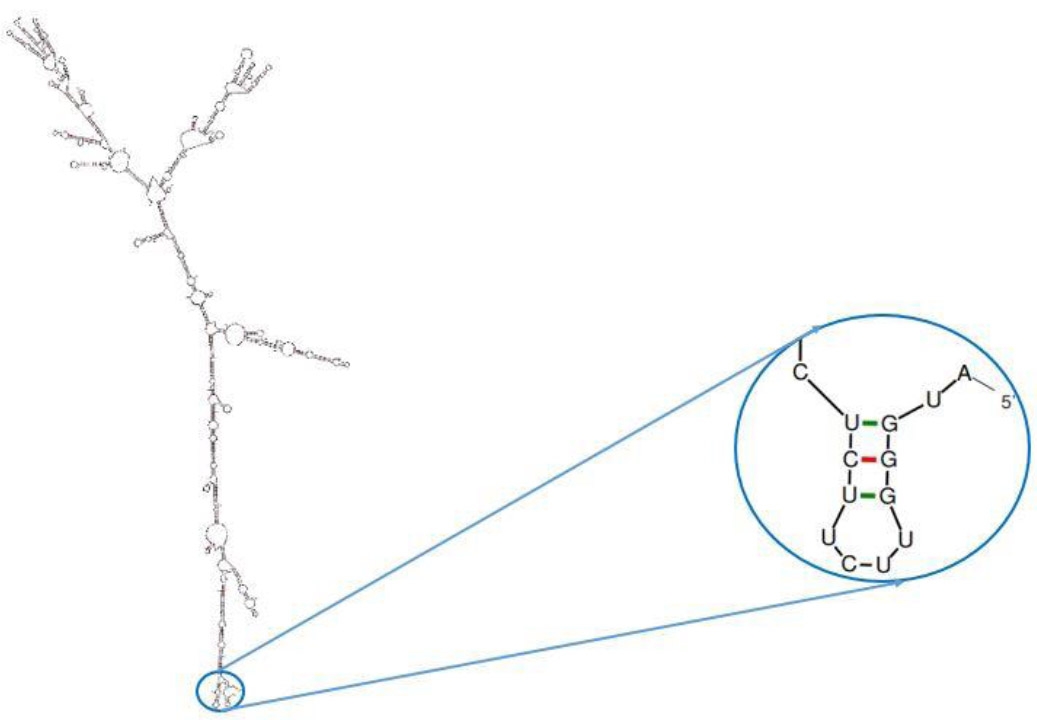
The prediction of the secondary structure of mRNA using mFold (http://unafold.rna.albany.edu/?q=mfold). The predicted structure lacks a hairpin and pseudo-knot at the 5′ site of the mRNA.
3.7. Antigenicity, Allergenicity, and Solubility Assessment
None of the chimeric proteins were predicted to be allergenic based on allergenicity assessments. Antigenicity scores were calculated as follows: 0.6717 for MRS1, 0.6785 for RMS1, 0.6694 for RSM1, 0.6713 for MRS2, 0.6779 for RMS2, and 0.6690 for RSM2. The solubility of the fusion protein was estimated at 0.581, indicating favorable characteristics for further development.
3.8. Prediction of Physicochemical Properties
The RSM1 protein had an isoelectric point of 9.35 and a MW of 53.81 kDa. The protein contained 57 negatively charged residues (Asp + Glu) and 75 positively charged residues (Arg + Lys). The half-life of RSM1 was approximately 30 hours in mammalian reticulocytes and extended beyond 20 hours in yeast and 10 hours in E. coli. Based on its instability index of 54.19, RSM1 was classified as unstable. Its GRAVY value was –0.639, and its aliphatic index was 68.66.
3.9. Optimization of the Codon
To enhance protein expression levels in E. coli, reverse translation was executed via the EBI server for codon optimization, ensuring that codons were optimized for the target organism to maximize expression efficiency.
3.10. Prediction of mRNA Secondary Structure
The mfold server was used to predict the optimal secondary structure of the mRNA, revealing a ΔG of –524.80 kcal/mol. The Minimum Free Energy (MFE) for the first 10 nucleotides at the 5′ end was –1.30 kcal/mol, suggesting that the formation of stable pseudoknot and hairpin structures is unlikely (Fig. 4).
4. DISCUSSION
The first step in successful vaccine development is antigen recognition, followed by identification of the parasite’s immunodominant epitopes [39]. T. gondii expresses a wide variety of antigenic epitopes, and antigen presentation varies among individuals. To address this complexity, recent vaccine development increasingly relies on in silico approaches that use bioinformatics tools to predict and select highly immunogenic epitopes. These computational methods enable the identification of epitope-rich regions across antigens, enabling more targeted vaccine design [40]. These methods can significantly reduce the time and cost of experimental testing [41]. In the in silico method, any antigen can be considered a vaccine candidate; however, our selection strategy prioritized antigenic diversity by including both surface (e.g., SAGs) and secretory (e.g., MICs, ROPs) antigens. Vaccines designed to elicit responses against multiple antigens typically offer broader protection compared to those based on a single antigen, which often lack sufficient cytotoxic T lymphocyte epitopes and fail to induce sterile immunity in acute or chronic toxoplasmosis [42-44]. Additionally, we selected antigens expressed across all three life stages of the parasite to ensure a more comprehensive immune response, as stage-specific antigens tend to provide limited protection [45]. T. gondii invades host cells through contact, gliding motility, moving junction (MJ) formation, and parasitophorous vacuole (PV) development [46-48]. SAGs of T. gondii are integrated within the plasma membrane via glycosylphosphatidylinositol anchors [49]. The MIC proteins are then released and spread over the parasite surface. These proteins are involved in the recognition and binding to host cell surfaces. Furthermore, the synergy and interaction between rhoptry neck and MIC proteins in the host plasma membrane stimulate gliding motility and formation of the MJ [50-54]. Following MJ formation, the parasite invades the host cell, forms a PV, and secretes ROPs, which are critical for PV development and host cell manipulation [55-57]. The mechanism of action of each of these antigens, ROPs, MICs, and SAGs, highlights their collective role in host cell invasion and underscores their contribution to T. gondii virulence. Previous studies have utilized various T. gondii antigens, individually or in combination, to develop DNA- or protein-based vaccines [53, 58-61]. For example, Li et al. employed a recombinant canine adenovirus (CAV-2) expressing the ROP18 gene, which induced strong Th1-skewed humoral and cellular responses in mice [61]. Petersen et al. reported that intramuscular vaccination with recombinant SAG1 plus alum triggered a Th2-biased response and resulted in limited survival [58]. Nabi et al. demonstrated enhanced antibody responses in mice immunized intranasally with rROP18-loaded nanospheres compared to other delivery methods [60]. Wang et al. found that a combined SAG1-MIC4 antigen provided greater protection than either antigen alone [53]. Additionally, multicomponent vaccines such as MIC1-4 and MIC1-4-6 elicited stronger immune responses, reduced brain parasite loads, and improved survival rates in mice [59]. In another study, a bioinformatics evaluation of the RMS protein, comprising MIC13, GRA1, and SAG1 antigens, suggested that this antigen could be a promising candidate for the development of a protective vaccine against T. gondii [62]. These findings underscore the superior efficacy of multigene vaccines over single-antigen approaches for toxoplasmosis prevention. Sequence conservation analyses further revealed that these antigens possess highly conserved regions across different T. gondii strains, supporting their potential as broad-spectrum vaccine targets. SAG1, for instance, is highly conserved among types I, II, and III strains, reinforcing its stability as a vaccine candidate [63]. ROP18 exhibits allele variation but retains core functional domains across strains, and its critical role in virulence suggests conserved regions are immunologically relevant [64]. MIC13, frequently included in multi-antigen vaccines alongside conserved antigens like SAG1 and ROP18, demonstrates conserved immunogenic potential [62, 65]. Together, these factors guided the prioritization of these antigens over others, aiming to maximize vaccine efficacy by targeting key molecules involved in parasite-host interactions. Based on this rationale, the current study focuses on designing a chimeric protein vaccine that contains B- and T-cell epitopes of ROP18, SAG1, and MIC13 antigens of T. gondii, and analyzing various aspects of this protein using different bioinformatics tools. A combination of three antigens (ROP18, SAG1, and MIC13) was used for the first time in the present study. On the other hand, T. gondii has a large number of antigens and antigenic epitopes. The most immunogenic epitopes of one antigen were selected using an in silico method, in combination with the most immunogenic epitopes of the other two antigens, which is a novel aspect of this study.
The study involved the construction of a chimeric protein composed of three specific antigens: ROP18, SAG1, and MIC13. During epitope selection, unstable regions and restriction sites were eliminated. CD8+ T cells that secrete interferon-γ play a crucial role in combating toxoplasmosis; however, the activation of B cells and the production of antibodies are equally vital for preventing the proliferation of the parasite within tissues during the chronic phase of the disease [66]. Therefore, in the selection of epitopes from these three antigens, epitope selection was based on the identification of fragments that are immunodominant in both B and T cells. Structurally, linkers used in multi-domain protein design are generally classified into flexible, rigid, and in vivo cleavable linkers [67]. Among these, rigid linkers are often more effective than flexible linkers for the separation of the functional domains [67]. One of the consequences of omitting a linker or using an inappropriate one is the misfolding of the chimeric protein [68], reduced protein expression [69], or the disruption of biological activity [70, 71]. In this study, the objective was to identify immunodominant fragments from these three antigens that would effectively stimulate both B and T cells. For this research, a rigid linker with the sequence A(EAAAK)nA was employed to connect the ROP18, SAG1, and MIC13 domains, with RSM1 chosen for its high antigenic potential among various configurations. This particular structure was notable for its low allergenic properties in addition to its elevated antigenicity. Antigenicity refers to the ability of a protein to be recognized by the immune system, and an adhesion found in the T. gondii proteome is likely to be antigenic. However, antigenicity alone was not sufficient to select a structure, and the tertiary structure of the protein was considered. Therefore, this structure should select final models based on the tertiary structure. I-TASSER reports up to five models that correspond to the five largest structure clusters. The C-score quantitatively measures the confidence of each model. The C-score is typically within the range of -5 to 2, where a higher C-score value signifies a model with high confidence, and vice versa. Among the modeled structures, RSM1 achieved the highest C-score, identifying it as the most suitable candidate for further investigation. The C-score is a valuable metric for evaluating the quality of predicted protein models, with higher values indicating greater confidence in the structural accuracy. In addition to this metric, structural validation was performed using a Ramachandran plot, which assesses the stereochemical quality of protein models and offers insight into their potential biological functionality [72]. RSM1 exhibited a favorable distribution of residues within the favored and allowed regions of the Ramachandran plot, with minimal representation in disallowed regions, reinforcing its classification as a high-quality model. Secondary structure analysis revealed that RSM1 consists of 36.08% alpha-helix, 14.23% extended strand, and 49.69% random coil. The presence of alpha helices and beta-turns is significant for maintaining protein stability and enhancing interactions with antibodies [73].
Additionally, when designing a vaccine, the physicochemical properties of the protein are fundamental. In this case, the aliphatic index was calculated to be 68.66, suggesting that the protein can maintain stability across a broad temperature range. However, with an instability index exceeding 40, the protein is expected to be unstable. Another important characteristic is the GRAVY score, where a negative GRAVY value suggests the protein is hydrophilic, which implies better interaction with water molecules in its environment [74]. The MW of the protein, a key factor for immune system activation, is also critical. Since an MW greater than 5 to 10 kDa is generally considered favorable for immunogenicity [33], RSM1, with its weight of 53.811 kDa, emerges as a promising antigen candidate. The isoelectric point is a key physicochemical property that plays a crucial role in assessing the solubility of proteins at specific pH levels. By determining the isoelectric point, it is possible to predict protein solubility. Typically, proteins with a pH of the solution equal to or near their isoelectric point tend to precipitate from solution [23]. Additionally, the half-life of a protein refers to the time required for half of the protein to be degraded following its synthesis within a cell. The ProtParam tool, which estimates half-life based on the N-terminal residue, may have limitations for certain analyses [24]. An important factor in codon optimization is the Codon Adaptation Index, which ranges from zero to one, and a value of one indicates that a gene uses synonymous codons for each amino acid with maximum frequency [75]. Poor codon adaptation can result in reduced or failed protein expression [24]. Additionally, mRNA stability is influenced by its MFE, as well as predictions of mRNA secondary structure, which can be performed with tools like mfold. According to mfold data, stable mRNA structures are generally more favorable for efficient translation and chimeric protein production in host systems. A primary limitation of this study is its reliance on in silico analyses based on computational predictions rather than experimental validation. While bioinformatics tools are powerful for predicting protein structures, epitopes, and antigenicity, the actual immunogenicity and biological activity of the RSM1 chimeric protein can only be confirmed through in vivo studies. B- and T-cell epitope prediction tools depend on existing databases and algorithms that may not fully capture all variables influencing immune responses; consequently, predicted epitopes might not accurately represent their immunogenic potential in biological systems. In addition, the prediction of T cell epitopes based on mouse-specific MHC alleles may limit the generalizability of the findings to other species, as immune responses can vary significantly across genetic backgrounds. To address these limitations and confirm the immunogenic potential of the designed vaccine, this study will proceed to experimental phases. The RSM1 gene will be cloned into a bacterial expression vector and expressed in E. coli. Following purification and quantification of the recombinant protein, immunization studies will be conducted in BALB/c mice. Humoral and cellular immune responses will be assessed using ELISA and cytokine profiling, while protective efficacy will be evaluated through challenge experiments and survival analyses. These steps will generate essential data regarding the safety, immunogenicity, and protective efficacy of the chimeric vaccine candidate.
CONCLUSION
In conclusion, this study employed in silico approaches to design a novel chimeric vaccine candidate, RSM1, by incorporating immunodominant epitopes derived from the key antigens ROP18, SAG1, and MIC13 from T. gondii. In silico analysis of the RSM1 construct suggested its potential as a promising vaccine construct for the development of a protective vaccine against T. gondii. Therefore, these findings underscore the utility of bioinformatics tools in vaccine development and enable the identification of epitopes that enhance immune responses. However, the findings from in silico analysis require validation through heterologous expression and subsequent in vivo experimentation.
AUTHORS' CONTRIBUTIONS
The authors confirm contribution to the paper as follows: T.N. conceived and designed the study protocol and served as the supervisor of this research. T.N. performed the bioinformatics analysis and drafted the manuscript. A.D., F.G., and A.M. critically revised the manuscript. All authors read and approved the final version of the manuscript.
LIST OF ABBREVIATIONS
| SAGs | = Surface Antigens |
| ROPs | = Rhoptry Proteins |
| MICs | = Microneme Proteins |
| GRAs | = Dense Granule Proteins |
| IEDB | = Immune Epitope Database |
| MHC | = Major Histocompatibility Complex |
| GOR | = Garnier-Osguthorpe-Robson |
| 3D | = Three-Dimensional |
| C-Score | = Confidence Score |
| MW | = Molecular Weight |
| GRAVY | = Grand Average of Hydropathicity |
| EBI | = European Bioinformatics Institute |
| MFE | = Minimum Free Energy |
ACKNOWLEDGEMENTS
The authors would like to thank the Infectious and Tropical Diseases Research Center, Dezful University of Medical Sciences, Dezful, Iran, for their support, cooperation, and assistance throughout the period of the study (IN&TR-403029-1403).

