All published articles of this journal are available on ScienceDirect.

Identification of Proteases: Carboxypeptidase and Aminopeptidase as Putative Virulence Factors of Fusarium solani Species Complex
Abstract
Background:
Fusarium keratitis accounts for around 50% of mycotic keratitis cases. Major virulence factors produced by keratopathogenic fungi are proteases.
Objective:
The aim of the current study was to identify proteases contributing to corneal pathogenicity of Fusarium species.
Methods:
Culture filtrates from fourteen Fusarium solani species complex (FSSC) isolates and three F. delphinoides isolates were evaluated for protease activity and gelatine zymography. Mass spectroscopy was carried out using a partially purified enzyme and total extracellular extract. Protease gene expression in an in-vitro condition and an ex-vivo goat corneal infection model was measured using qRT-PCR. Specific activity was observed in a wide range and at a broad pH range; and isolates Cs1 (maximum) and Cc50 (minimum) were selected for the infection model.
Results:
Gene expression in in-vitro condition showed the highest fold change for proteases (C7YY94, C7Z7U2 and C7Z6W1) while in an ex-vivo infection highest fold change was seen for proteases (C7Z6W1, C7YQJ2 and C7Z7U2); in decreasing order, respectively. Expression of aminopeptidase (C7Z6W1) was 50-fold higher in the infected cornea in both isolates (Cs1 and Cc50); while expression of carboxypeptidase (C7YVF3) was 15-fold higher only in isolate Cs1. Corneal histology showed less penetration of Cc50 than Cs1 into the stroma. Mass spectrometry showed the presence of carboxypeptidase (C7YVF3) and tripeptidyl amino peptidase.
Conclusion:
It can be concluded that clinical isolates of FSSC produce varying amounts of proteases and differ in specific activity and gene expression in both conditions (in vitro and ex vivo). Carboxypeptidase and aminopeptidase contribute to the pathogenic potential of Fusarium solani species complex.
1. INTRODUCTION
Fungal keratitis is a devastating eye infection and is classified as an ophthalmological emergency. Patients with fungal keratitis undergo surgical intervention, like corneal transplantation, enucleation, or suffer deterioration of vision as a result of drug treatment failure [1]. The incidence of this dis-order has increased over the last 30 years. Keratitis causing fungi have gained increasing importance in Asian nations and the contribution is almost half of the keratitis cases reported worldwide [2, 3]. In Asian nations, among all microbial keratitis cases, fungal keratitis contributes to nearly 40% of the total cases [4, 5]. Corneal epithelial defects caused by trauma usually involve plant debris as the major predisposing factor. However, in urban regions with moderate temperature, increased use of contact lens associated keratitis has become a problem [6-9].
Fusarium infections are difficult to treat because of increased antifungal resistance of Fusarium species [9-11]. Pathogenic Fusarium species show resistance to a wide range of antifungals in vitro. with high variation at species level [12-14]. In clinical infections, the most commonly observed pathogenic strains belong to FSSC and the minimum inhibitory concentration (MIC) of FSSC species against antifungals is very high [15]. Many studies report the prevalence of Aspergillus spp., followed by Candida and Fusarium spp. However, there is a shift in this prevalence in recent studies where Fusarium is now reported as the most common fungal pathogen for keratitis. A 5-year retroactive review of mycotic keratitis in Malaysia showed an increasing prevalence of fungal keratitis. Between 2007-2011, the percentage of mycotic keratitis incidences increased from 12.50% to 28.57% [16, 17].
Several species of Fusarium are known to cause keratitis and keratitis by members of this genus comprise higher than 50% of all mycotic keratitis cases. Fungal keratitis caused by several species of Fusarium, including the Fusarium solani species complex (FSSC), F. avenascus, F. moniliforme (F. verticilliodes), F. oxysporum, F. dimerum, F. poae, F. fujikuroi, F. chlamydosporum, F. incarnatum-equiseti species complexes and F. proliferatum [18-20]. FSSC is the most common in causing mycotic keratitis [1]. Nearly 50% of all fusariosis cases are attributed to members of the FSSC [21].
A determinant of pathogenicity is a virulence factor. Virulence factor is a microbial component that damages the host [22]. Pathogenic fungi preserve many virulence factors that help them grow in unfavourable environment imposed by the host and lead to the development of the disease. Proteases and lipases are hydrolysing enzymes which are extremely important virulence factors retained by pathogens [23]. Extracellular proteinases of pathogens may play a role in mucosal surface adhesion and survival, host tissue invasion and immunoglobulin digestion. Fungi colonized on mucosal surface, secrete proteases which helps it to penetrate tissue of host during disseminated infections [24]. Extracellularly secreted proteins are primary proteins which interact with host at first and play role infection development [25].
Our aim is to quantify the proteases secreted by keratitis causing Fusarium (FSSC) isolates, to characterize them and to study their expression profile in an in-vitro condition and in an ex-vivo goat corneal infection model.
2. MATERIALS AND METHODS
2.1. Fungal Culture
The present work was carried out on 17 clinical isolates of Fusarium spp. of which 14 were FSSC isolates (CSH1, CSH2, CSH3, CSH5, CSH6, CSH7, CSH8, CSH9, CSH10, CSH11, Cs1, Cs2, Cc50 and Cc240), three were F. delphinoides isolates (Cc26, Cc119 and CSH4) which were earlier obtained from keratitis patients. All isolates were identified using the internally transcribed spacer (ITS) and translational elongation factor (TEF) region sequencing. The Fusarium spp. (CSH isolates) included in the study were isolated from corneal scrapings of mycotic keratitis patients which were provided by Jhaveri Microbiology Centre, L V Prasad Eye Institute, Hyderabad.
2.2. Growth of Fungi in an in vitro Condition
Potato Dextrose Agar (PDA) (Himedia Laboratories, India) was used to grow all isolates for seven days and then approximately 10mm disc was cut from the PDA plate and inoculated in 100ml of sterile PDB (Himedia Laboratories, India) and grown for eight days at 30±2 ºC in static condition.
2.3. Proteases Extraction and Precipitation from an in vitro Culture
At the end of 8th day, fungal culture filtrate was separated from mycelia using sterile Whatman No. 1 filter paper (Merck, USA) and collected in a sterile bottle and precipitated using chilled acetone (Merck, USA). Chilled acetone (4 ml) was slowly dropped to fungal culture filtrate (1 ml) and kept at -20ºC for precipitation for 24 hrs. After 24 hrs, the precipitate was collected by centrifugation at 5000xg for 15 min at 4ºC. Precipitate was dissolved in Phosphate Buffered Saline (PBS) and stored at 4ºC until further use for azocaesin assay and gelatine zymography. Bradford assay was used to assess protein concentration of concentrated extract using Bovine Serum Albumin (Sigma-Aldrich, USA) as standard.
2.4. Protease Specific Activity Estimation by Azocasein Assay
Azocasein (Sigma-Aldrich, USA) (5 mg/ml) was dissolved in assay buffer containing 50mM Tris (Sigma-Aldrich, USA) (pH 7.4), 0.2M NaCl (Himedia Laboratories, India), 5mM CaCl2 (Himedia Laboratories, India) 0.01% sodium azide (Himedia Laboratories, India) and 0.05% Brij 35 (Himedia Laboratories, India). Azocasein solution (400μl) was mixed with concentrated extract (100μl) and incubated at 37°C for 90 min. The reactions were stopped by the addition of 150μl of 20% trichloroacetic acid (TCA) (Sigma-Aldrich, USA) and allowed to stand for 30 min at RT. Supernatant was collected by centrifugation at 8,000xg for 3-4 minutes. 500μl of supernatant was mixed with 1M NaOH (Himedia Laboratories, India) (500μl). The released azo dye was estimated by measuring absorbance at 436nm. Enzyme activity (1 unit) was defined as an increase of 0.1 absorption unit after incubation for 1 hour. Specific activity was calculated as Units/ mg of protein.
2.5. Characterization of Proteases
For protease characterization, primarily, the effect of pH and inhibitors was studied on crude enzyme extract.
2.5.1. Effect of pH
To study the effect of pH on enzyme activity, enzyme concentrate was incubated with azocasein solution of respective pH. Azocasein assay was carried out at pH ranging from acidic to basic i.e. pH 3, pH 5, pH 7.4, pH 8 and pH 10.
2.5.2. Effect of Enzyme Inhibitors
To understand the type of enzyme produced, crude enzyme extract was incubated with specific inhibitors and azocasein assay was carried out. PMSF (Sigma Aldrich, USA) (inhibitor of serine protease) (5mM), Pepstatin A (Sigma Aldrich, USA) (inhibitor of aspartyl proteases) (10μM), and EDTA (Himedia Laboratories, India) (inhibitor of metalloprotease) (10mM) were used to identify the type of proteases present in Fusarium isolates. Crude enzyme extracts were incubated with inhibitor for 1 hour (at 37 ºC temperature) and the same inhibitor concentration was used for azocasein assay.
2.6. Gelatine Zymography
Zymography was carried out using gelatine (Sigma Aldrich, USA) as a substrate. Twelve percent Sodium Dodecyl Sulphate - polyacrylamide (Merck, USA) gel (SDS-PAGE) was prepared with gelatine (0.1%). 0.2 Units of the enzyme were mixed with 6X gel loading dye and electrophoresed. After electrophoresis, the gel was washed with distilled water (D/W) and incubated in a buffer containing 50mM Tris (Himedia Laboratories India), 5mM CaCl2 (Himedia Laboratories, India, 1μM ZnCl2 (Himedia Laboratories India) and 2.5% Triton –X 100 (Himedia Laboratories, India) for 1 hour. The gel was washed twice with D/W and incubated in an incubation buffer for overnight. After incubation, gel was again washed with D/W and stained with Commassie Brilliant blue R-250 (Himedia Laboratories, India) (0.5%) overnight. Destaining of gel was carried out using methanol (Merck, USA): water: acetic acid (Merck, USA), 4:5:1 (v/v). The enzyme activity was observed by bands of substrate degradation against blue background. Zymography was also carried out in the presence of inhibitors. The enzyme samples were preincubated with the previously mentioned inhibitor concentration for 1 hour and then subjected to electrophoresis.
2.7. Purification of Protease
2.7.1. Affinity Column Chromatography
Casein agarose (Sigma-Aldrich, USA) column was used as matrix. 10ml syringe without a needle was used as a column and was packed with the matrix. The column was pre-sealed with glass wool at the base. Casein agarose (5.0ml) was poured slowly, avoiding the presence of any air bubble in the matrix and was washed with 50mM phosphate buffer (pH 8.0) so that all the salt in the suspension of casein agarose is washed out. The whole column was equilibrated by passing 2-3 bed volume of 50mM phosphate buffer (pH 8.0). Crude enzyme extract (isolate Cs1) was loaded into the column (2ml). It was allowed to bind and was washed with 20 bed volume of 5mM phosphate buffer (pH 8.0) so that no unbound protein remains in the matrix. After washing, elution was done against a concentration 0.1M – 1.0M NaCl. Elution of protein was done at a flow rate of 0.25ml/min. Fractions were analysed for activity using azocasein assay and total protein concentration was estimated using Bradford assay.
2.8. Selection of Protease Genes and Primer Designing for Quantitative Real Time Polymerase Chain Reaction
Primer designing for protease genes was done for Nectria haematococca (telomorph of F. solani), as whole genome data is available for N. haematococca. UniProt KB was used to find out proteases present in N. haematococca. Proteases ranging from 50 – 100 kDa were selected. The gene sequence for these proteases was obtained from Ensembl Fungi and National centre for Bioinformatics (NCBI). Elongation factor 1 alpha (EF 1 alpha) was used as housekeeping gene. Primers were designed using primer designing tool NCBI Primer BLAST. The quality and properties of primers were checked using OligoCalc (oligonucleotide property calculator). Primers were synthesised commercially by Eurofins, India.
2.9. RNA Isolation
Dried fungal mycelial mat (100 ng) was homogenised in 1 ml of RNA Iso plus reagent (TAKARA, Japan) and kept at room temperature (RT) for 5 min. Chloroform (0.2ml) (Merck, USA) was added, swirled for 15- 30s and kept at RT for 2-3 min followed by centrifugation at 4ºC for 15 min 12,000xg. After centrifugation 3 layers were separated, the uppermost layer which contained RNA was taken and 0.5ml of isopropanol (Merck, USA) was added. Contents were mixed by gently inverting the tubes and kept at RT for 10-15 min. Centrifugation was carried out at 12,000xg at 4ºC for 10 min. Supernatant was discarded and the pellet was washed with 70% ethanol twice, followed by centrifugation at 7,500xg at 4ºC for 5 min. The pellet was air-dried for 5-10 min and then dissolved in nuclease free water. Quality of RNA was checked by agarose gel electrophoresis using 2% agarose gel and by taking ratio of 260/280 nm and RNA quantification was done by measuring absorbance at 260 nm using spectrophotometer (Multiscan Go, Thermo fisher scientific, USA). After quantification RNA was treated with DNase (Rosche, USA) to remove possible DNA contamination. Once RNA was free of DNA contamination, it was stored at -20ºC till further use.
2.10. cDNA Synthesis
For cDNA synthesis; Primerscript 1st strand cDNA synthesis kit (TAKARA, Japan) was used. Kit was provided with random 6-mer primer, oligo dT primer, dNTPs, 5X primer script buffer, Reverse transcriptase, RNase inhibitor and nuclease free water. Five microgram of RNA was taken as a template. 2μl of random 6-mer primer, 1μl of oligo dT primer and 1μl of dNTPs were added to it. Final volume was made 10μl with nuclease free water. Reaction was carried out in PCR thermocycler (Bio-Rad, USA). PCR reaction involved incubation at 30ºC for 10 min followed by 65ºC for 5 min. Reaction was stopped and PCR tube was placed on ice for 5 min. Then, 4μl of 5X primer script buffer, 1μl of Reverse transcriptase and 0.5μl of RNase inhibitor were added. Final volume was made up 20μl with nuclease free water. Reverse transcription was carried out as follows: 50ºC for 1 hour, 75ºC for 15 min and halt at 4ºC. cDNA was stored at -20ºC till further use.
2.11. Quantitative RT-PCR (qRT-PCR)
Real time PCR was carried out using Bio-Rad CFX Manager thermocycler. cDNA was used as template. SyBr Green premix (TAKARA, Japan) was used as fluorescent dye for real time analysis. For the amplification of protease gene, respective primers (Table 1) were added to PCR reaction mixture. Reaction contained 2μl of cDNA template, 10pmol of forward and reverse primer of respective gene, 10μl of SyBR premix and final volume was set 20μl using dH2O. Reaction mixture was added to real time PCR plates (Thermo Scientific, USA) and plates were sealed with a sealant (Thermo Scientific, USA). The PCR reaction involved the following steps: 1 cycle of 95ºC for 3 min, 40 cycles of 95ºC for 30s, 55ºC for 30s and 72ºC for 30s. Plate read was at the end of each cycle. Final extension was carried out at 72ºC for 5 min. Melt curve was set at 65ºC-95ºC with an increment of 0.5ºC for 5s followed by plate read at every step. Amplified product was confirmed on 2% agarose gel. The fold change was calculated by using ΔΔCT method and MTCC 2935 was used as a reference strain.
2.12. Identification of Protease Using High Resolution Liquid Chromatography- Mass Spectrometry (HRLC-MS/MS)
Crude enzyme extract (acetone precipitated) and partially purified enzyme (obtained from column chromatography) were used for identification using MS/MS.
2.12.1. Sample Preparation
Crude enzyme extract of isolate Cs1 (FSSC) was concentrated by using 10 Kilo Dalton Molecular Weight (kDa M.W.) cut-off column (Merck, USA). Protein concentration of the obtained crude extract was measured using Bradford assay. The column purified fraction was concentrated by acetone precipitation method. 50µg of samples were taken and final volume was made up to 10µl with 25mM ammonium bicarbonate buffer. Then 20mM DTT was added to carry out the reduction. The samples were incubated at 56˚C for 1 hour in dry bath. Alkylation was carried out using Iodoacetamide and samples were incubated at RT for 30 min. Samples were then digested with trypsin in ratio of 1:30 (v/v). The sample was incubated at 37˚C for 16-18 hours. The samples were dried under vacuum. 20µl of 0.1% formic acid was added to the dried samples. Samples were de-salted with 50% acetonitrile (ACN), followed by 80% ACN C18 RP column was activated by using 100% ACN. Column was washed with 0.1% formic acid. The samples were again solubilised in 0.1% formic acid and passed through the column. Column was eluted using 50% ACN, followed by 80% ACN. The obtained fraction was then dried under a vacuum. The dried samples were re-suspended in 0.1% formic acid and used for MS/MS analysis using Q-Exactive Plus Biopharma-High Resolution Orbitrap (Thermo Scientific, USA).
| No | Uniprot ID | Name | MW (kDa) | Putative function | Primer |
|---|---|---|---|---|---|
| 1 | C7Z0E6 | Uncharacterized protein | 95.93 | serine-type endopeptidase activity | F:CTGGCGCAGGGAGGTAA R:TGGTTGAGGAGCGATATCCAT |
| 2 | C7ZFW9 | Uncharacterized protein | 97.81 | serine-type endopeptidase activity | F:CGAGTTTGAGGATGGAACCAA R:ATTCCTCCTCCTGGAGGGTAT |
| 3 | C7Z7U2 | Uncharacterized protein | 94.29 | serine-type endopeptidase activity | F:GTTGACAAGTTGCGAGCTGA R:TGCGTTCTTGCCGTTGTA |
| 4 | C7ZNV5 | Predicted protein | 92.61 | serine-type endopeptidase activity | F:AAGGCCGAAGGTTATGTCGA R:ATGGCTTCACCCCACAGCTT |
| 5 | C7YY94 | Uncharacterized protein | 94.32 | serine-type endopeptidase activity | F:TGCTGGAGAATCTTGATGCT R:TTGCCCTCTTTTAGAAGGGA |
| 6 | C7Z7Y4 | Palmitoyl transferase | 78.37 | Palmitoyl transferase activity | F:TGGGCCGCAATTAACAACCAG R:GGTGGAGAAGAAGCACGAG |
| 7 | C7YQJ2 | Carboxypeptidase Y homolog A | 60.16 | serine-type carboxypeptidase activity | F:GTGCTGGGCGCTGCGTCGT R:ACGTGATCCCAGTGGTTGT |
| 8 | C7YVF3 | Carboxypeptidase | 62.13 | serine-type carboxypeptidase activity | F:CCAGGCAGCTTTCAACCGGG R:CTCTGGACTCATCCAGGGT |
| 9 | C7YPA2 | Uncharacterized protein | 82.66 | serine-type endopeptidase activity | F:GTTTCAAAGAGTTCGAGTGC R:GACAGTGGCACCTTTGCCTCGT |
| 10 | C7Z8P9 | Uncharacterized protein | 65.65 | serine-type endopeptidase activity | F:ATGAAAGCGATATCACTCA R:GGCTTCCTTCTGGGTGA |
| 11 | C7YSA1 | Uncharacterized protein | 66.47 | serine-type endopeptidase activity | F:ATGCGGCCTTTGCTCGCGCTGA R:GCAAGACCTGTCGTGGTTGA |
| 12 | C7Z436 | Uncharacterized protein | 78.97 | serine-type peptidase activity | F:ATGCGGCCTTTGCTCGCGCTGA R:GTCCTTGAGGGCGTCGACTGTAGA |
| 13 | C7YSV9 | Uncharacterized protein | 56.83 | serine-type endopeptidase activity | F:GCCGTTTCTGAGTACAAGTGC R:GTTGCCATCCTCATCAGCATCGC |
| 14 | C7Z6W1 | Leucine aminopeptidase 1 | 43.54 | Aminopeptidase activity | F:AAGAACCTCGAGAAGAAGAAC R:CATCGTCATCGGCGCCAGGAGC |
| House-keeping gene | |||||
| 1 | EF1 alpha | Elongation factor 1 alpha | F: TTCAAGTGGGCGATGCTCTT R: AGTTGATGGGGTCTGCTGTG |
||
kDa – Kilo Dalton
2.12.2. MS/MS Parameters
A capillary column (C18, 2 μm, 100 Å, 75 μm × 50 cm, Thermo scientific, USA) was used for the separation of peptides. Sample was loaded first onto a pre-column from an auto-sampler at maximum pressure of 1000 bar. The sample was then analysed using an analytical column with flow rate of 300 nl/min (95% ACN in 0.1% FA from 5% - 10% for 5 min, 10% - 50% for 45 min, 50% to 95% for 5 min and kept at 100% for 5 min). Samples were analysed in positive mode of electrospray ionization. Full scan mass spectra were obtained over m/z of 350–2000 Da with a frequency of 1 spectrum per sec.
2.12.3. Data Analysis
The raw data were analysed using Proteome Discoverer 2.1 (Thermo Scientific, USA) using inbuilt SequestHT algorithm and Mascot (Matrix Science, London, UK). Following parameters were used for database search. 10 ppm peptide tolerance and 0.60-0.80 Da fragment tolerance and two missed cleavages. Fixed modification was Cysteine carbamidomethylation and variable modifications were N-terminal acetylation, methionine oxidation and phosphorylation (S, T, Y). The percolator algorithm was used for the processing of peptide spectrum matches (PSMs) from Mascot and SequestHT.
2.13. Ex vivo Caprine (goat) Cornea Infection Model of F. solani
Ex vivo corneal infection model was previously developed in the lab (Madhu et al., 2018). It was performed with few modifications. Briefly, goat eyeballs were collected from an abattoir in a sterile beaker. Surface sterilization was carried out with 2.5% povidone iodine (Cipla, India) for 5 by gentamycin (Talent health care, India) treatment for 15 min. The dissection of eyeball was carried out with the help of sterile corneal scissors and cornea was separated. Artificial corneal buttons were made from 1% agarose (Merck, USA) prepared in Dulbecco’s Modified Eagle medium (DMEM) (Gibco, Thermo Fisher, USA) and poured in sterile semicircle stainless steel beads. These beads were placed in sterile 12 well tissue culture plate (Eppendrof, USA). Dissected corneas were washed with sterile PBS and placed on artificial corneal beads. DMEM containing 75 μg/ml streptomycin (Gibco, Thermo Fisher, USA), 35 μg/ml gentamycin, 100 I.U./ml penicillin (Gibco, Thermo Fisher, USA) and 10% FBS was added to each well. Cornea was scratched gently from the epithelial surface and infected with 104 spores of F.solani. The plate was incubated at 37˚C and 5% CO2 concentration in CO2 incubator. Media was changed after every 6 hours and was estimated for protease activity using azocasein assay. Histology of infected corneas at 8th day was carried by method of Madhu et al., 2018. Briefly, the infected corneas were fixed with 2% buffered paraformaldehyde solution. These corneas were paraffinized and cut into the section of 5μm thickness and placed on silane coated slides followed by deparaffinization and Hematoxylin-Eosin (H-E) staining.
3. RESULTS
3.1. Fusarium Proteases - In vitro
The description about Fusarium proteases in vitro are as follows:
3.1.1. Specific Activity of Proteases
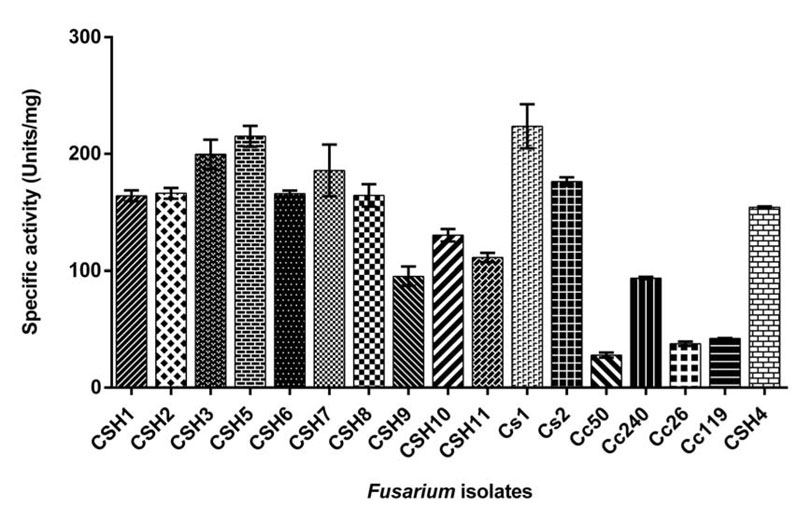
Azocasein assay was carried out for specific activity estimation in all isolates. (Fig. 1) shows the specific activity of all Fusarium isolates. The maximum activity in FSSC isolates was 223.68±18.83 units/mg (Cs1) and minimum activity was 30.05±0.73 units/mg (Cc50). In F. delphinoides isolates, Cc26, Cc119 and CSH4 had specific activity of 37.65±1.92 units/mg, 42.24±0.24 units/mg and 154.15±0.80 units/mg, respectively.
3.1.2. Characterization of Protease
Effect of pH and inhibitors was studied for protease characterization.
3.1.2.1. Effect of pH
For all Fusarium isolates, maximum activity was observed at pH 7.4. This indicates the majority of proteases being produced by Fusarium isolates are neutral proteases. Supplementary Table 1 shows the activity of proteases at different pH from all Fusarium isolates.
3.1.2.2. Effect of Inhibitors
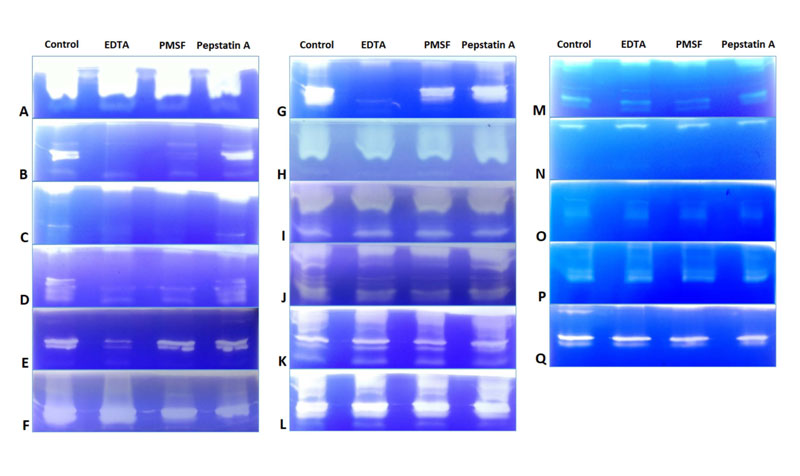
Effect of inhibitors was studied using azocasein assay and gelatine zymography. In azocasein assay, specific activity of control (without inhibitors) was considered as 100% and with respect to that the % residual activity was calculated for all isolates in presence of respective inhibitors. Table 2 shows % residual activity of crude precipitated enzyme in presence of EDTA (10mM), PMSF (5mM) and Pepstatin A (10μM). Initially the inhibitor concentration used was EDTA (1mM), PMSF (5mM) and Pepstatin A (1μM), but no inhibition was seen in azocasein assay and gelatine zymography (data not shown). In gelatine zymography, control lane showed maximum activity by clear band of substrate digestion and inhibition of activity was observed in lanes in which enzyme pre incubated with inhibitors [EDTA (10mM), PMSF (5mM) and Pepstatin A (10μM)] were loaded. All three types of proteases were detected in all Fusarium isolates. In FSSC isolates, inhibition was seen majorly by Pepstatin A and EDTA followed by PMSF in azocasein assay while in case of gelatine zymography, major inhibition was seen majorly by EDTA and PMSF followed by Pepstain A. In some of the FSSC isolates no inhibition was detected in zymography which were CSH1, CSH9, CSH10, Cs1, Cs2 & Cc240. Few isolates were inhibited by both EDTA and PMSF which were CSH2, CSH3, CSH5 & CSH11. Isolates which were inhibited by EDTA only were CSH6 and CSH8. CSH7 showed inhibition with PMSF & and Pepstatin A. Cc50 showed inhibition only with Pepstatin A (Fig. 2A to N). In F. delphinoides isolates (Cc26, Cc119 and CSH4) showed inhibition only by EDTA in azocasein assay, but no inhibition was detected in zymography (Fig. 2O, P and Q).
3.1.3. Quantitative RT-PCR (qRT-PCR) in vitro
Quantitative RT-PCR was carried out only for FSSC isolates only as mentioned earlier. Among all FSSC isolates, of 14 selected proteases genes (Table 1) expression of seven genes (C7Z7U2, C7Z7NV5, C7YQJ2, C7YVF3, C7YY94, C7Z436 and C7Z6W1) was detected using qRT-PCR. Supplementary Table 2 shows fold change in respective genes in all FSSC isolates. Among these seven genes, maximum expression was seen in C7YY94 followed by C7Z7U2 and C7Z6W1. On the basis of expression studies and specific activity assay two isolates which were selected for an ex vivo infection model were Cc50 and Cs1 with minimum and maximum specific activity, respectively.
3.1.4. Proteases Identification by HRLC-MS/MS
FSSC isolate Cs1 was used for protein identification directly from crude extract by Mass Spectrometry. Also, crude extract of Cs1 was used for purification by affinity chromatography. After purification from column, fraction 15 showed maximum specific activity (data not shown). This fraction was used for protein identification. Table 3 shows proteases identified from exoproteome of crude extract of Cs1 and from column purified fraction 15. The peptide sequences of Cs1 was matching with mitochondrial intermediate peptidase (C. albicans) (Accession no: Q59RK9), Pro-apoptotic serine protease (S. cereviseae) (Accession no: A6ZRW1), carboxypeptidase (N. haematococca) (Accession no: C7YVF3), aminopeptidase (Pseudomonas) (Accession no: C3K6G5), Metallopeptidase (Pseudomonas) (Accession no: A0A0C5CJR8), Predicted protein (having peptidase activity) (N. haematococca) (Accession no: C7YVP3). The column purified fraction shows the similarity with tripeptidyl amino peptidase (F. langsethiae) (Accession no: A0A0M9ESD1).
3.2. Fusarium Proteases - Ex Vivo
3.2.1. Specific activity of proteases in an ex vivo caprine (goat) corneal infection model of FSSC isolates
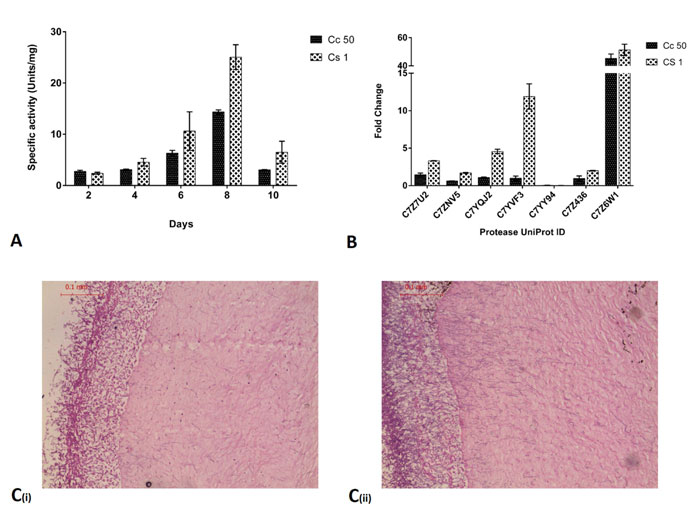
(Fig. 3A) shows the specific activity of proteases in an ex vivo goat corneal infection model of Cc50 and Cs1 from spent medium at 2nd, 4th, 6th, 8th and 10th day. The activity was increased upon progression of infection and was maximum at 8th day. Protease activity in Cs1 (25.05±2.424 units/mg) infected cornea was slightly higher than Cc50 (14.348±0.396 units/mg) at 8th day. RNA isolation from infected cornea was carried out on 8th day from both isolates.
| Isolate | % Residual activity | ||
|---|---|---|---|
| EDTA (10mM) | PMSF (5mM) | Pep A (10μM) | |
| FSSC isolates | |||
| CSH1 | 73.86 ± 6.25 | 94.31 ± 12.90 | 79.16 ± 3.28 |
| CSH2 | 80.57 ± 1.89 | 108.26 ± 5.85 | 85.12 ± 3.98 |
| CSH3 | 131.22 ± 3.69 | 104.91 ± 1.21 | 91.92 ± 5.79 |
| CSH5 | 79.62 ± 1.60 | 87.03 ± 3.20 | 94.44 ± 2.77 |
| CSH6 | 71.57 ± 1.75 | 73.09 ± 2.63 | 88.83 ± 2.32 |
| CSH7 | 93.86 ± 4.25 | 87.11 ± 2.81 | 95.09 ± 2.12 |
| CSH8 | 76.35 ± 2.05 | 145.10 ± 2.44 | 120.38 ± 3.29 |
| CSH9 | 59.77 ± 7.17 | 145.97 ± 5.26 | 75.86 ± 8.67 |
| CSH10 | 70.58 ± 5.63 | 128.87 ± 2.45 | 97.86 ± 0.92 |
| CSH11 | 42.26 ± 1.19 | 79.38 ± 7.01 | 69.07 ± 1.19 |
| Cs1 | 93.93 ± 3.14 | 152.12 ± 7.92 | 83.63 ± 2.77 |
| Cs2 | 87.30 ± 3.51 | 113.19 ± 6.15 | 83.75 ± 7.51 |
| Cc50 | 104.76 ± 7.14 | 142.85 ± 17.97 | 54.76 ± 7.14 |
| Cc240 | 96.92 ± 0.36 | 98.46 ± 0.72 | 99.48 ± 0.36 |
| F. delphinoides isolates | |||
| Cc26 | 46.85 ± 4.42 | 56.52 ± 2.21 | 59.90 ± 9.20 |
| Cc119 | 28.49 ± 1.23 | 49.73 ± 4.65 | 33.60 ± 4.90 |
| CSH4 | 78.88 ± 3.20 | 93.70 ± 1.11 | 84.81 ± 3.57 |
PMSF - Phenylmethylsulfonyl fluoride
Pep A - Pepstatin A
| No. | Function | pI | M.W. (kDa) |
Nearest informative homologue given by BlastP Accession [organism] |
Nearest informative homologue given by UniprotKB Accession [organism] |
|---|---|---|---|---|---|
| Proteases identified in exoproteome | |||||
| 1 | Mitochondrial intermediate peptidase | 6.44 | 89.3 | 3646074 (Candida albicans) |
Q59RK9 (C. albicans) |
| 2 | Pro-apoptotic serine protease | 5.96 | 110.8 | 125863594 (Saccharomyces cerevisiae) |
A6ZRW1 (S. cerevisiae) |
| 3 | Carboxypeptidase | 5.63 | 62.1 | 9675077 (Nectria haematococca) |
C7YVF3 (N. haematococca) |
| 4 | Probable cytosol aminopeptidase | 7.66 | 52.4 | 229359445 (Pseudomonas fluorescens) |
C3K6G5 (P. fluorescens) |
| 5 | Metallopeptidase AprA | 4.88 | 49.4 | 757867863 (P. panacis) |
A0A0C5CJR8 (P. panacis) |
| 6 | Predicted protein | 5.53 | 58.7 | 9677116 (N. haematococca) |
C7YVP3 (N. haematococca) |
| Protease identified after purification by column chromatography | |||||
| 7 | Tripeptidyl aminopeptidase | 5.23 | 58.0 | 927755098 (Fusarium langsethiae) |
A0A0M9ESD1 (F. langsethiae) |
M.W. - Molecular weight
kDa - Kilo Dalton
3.2.2. Quantitative RT-PCR (qRT-PCR) in an ex vivo Corneal Infection Model of FSSC Isolates
(Fig. 3B) shows fold change of C7Z7U2, C7Z7NV5, C7YQJ2, C7YVF3, C7YY94, C7Z436 and C7Z6W1 in Cc50 and Cs1. In an ex vivo infection condition, among all seven expressed genes, C7Z6W1 showed the highest fold change followed by C7YQJ2 and C7Z7U2. Protease (C7YVF3) showed higher expression in Cs1 compared to Cc50. Other genes also showed upregulation during ex vivo infection.
3.2.3. Histology of an ex-vivo Infected Cornea
(Fig. 3C) shows the histology of infected cornea where Cc50 shows less hyphal penetration into stroma compared to Cs1. Also, the growth of fungi on 8th day is less in Cc50 compared to Cs1 which can be seen in outer layer. The epithelial layer has also been completely degraded in both cases.
4. DISCUSSION
Role of proteases from fungi causing keratitis has been reported in many studies [26-29]. However, the characterization and identification of these proteases in Fusarium keratitis are lacking. Proteases from keratopathogenic Fusarium spp. were documented in our previous reports in an in vitro condition [30] and during corneal infection (an ex vivo explant model) [31]. In the present study, using the MS/MS identification and quantitative RT-PCR we attempt to identify the possible proteases that are involved in corneal pathogenesis of Fusarium spp.
Initially in the present work, an in vitro characterization and zymography was done to narrow down the kind and number of proteases produced by FSSC. All, FSSC isolates in the present work produced more than one kind of protease and it was apparent that serine and metalloproteases are among the important ones in FSSC. Based on this initial information, protease genes ranging from 50–100 kDa were selected from the whole genome sequence of N. haematococca to study the expression of these genes in both, an in vitro and an ex vivo explant infection condition. We found four interesting findings in the present study: (i) different protease genes show expression during an in vitro growth and an ex vivo corneal explant conditions. For e.g. proteases (C7YY94, C7Z7U2, C7Z6W1) expressed during an in vitro growth while, proteases (C7Z6W1, C7YVF3) expressed during ex vivo corneal explant infection, (ii) carboxypeptidase (C7YVF3) was identified in HR-LCMS/MS and it was also highly expressed in the corneal infection, (iii) aminopeptidase (C7Z6W1) showed around 50-fold upregulation during corneal infection. We could not verify the presence of aminopeptidase (C7Z6W1) in mass spectrometry analysis but, another tripeptidyl aminopeptidase (TPP) that showed homology to a TPP from F. langsethiae (A0A0M9ESD1) was detected and, (iv) we also found another two serine peptidases which showed homology to Q59RK9 (C. albicans) and A6ZRW1 (S. cerevisiae) using mass spectrometry. When these proteins where blasted against the N. haematococca genome; proteins matching with 49.5% and 32.47% similarity were found which are predicted and yet to be annotated.
The first report on the role of proteases in collagen destruction was shown in rabbit keratitis [32]. Later, characterization of proteases from A. flavus and F. solani was carried out using an in vivo in the rabbit model of keratitis. Researchers found out that 200kDa, 92kDa and 58kDa gelatinases were found in the infected cornea with both A. flavus and F. solani. Also, 65kDa protease was found in all infected and non-infected eyes. These gelatinases showed inhibition with EDTA and were considered as metalloproteases [27, 33]. Our results also corroborate with their findings as proteases of molecular weights of around 100kDa and 60-50 kDa were found in zymography. We believe that the high molecular weight band found in zymography may correspond to the proteases C7Z7U2/C7ZNV5 and the low molecular weight bands may correspond to proteases – C7YVF3 and C7Z6W1. Our study warrants the need to characterize these unannotated genes using gene deletions and over expression studies to validate the role of these proteases as a promising virulence factor. Earlier reports from non-keratopathogenic Fusarium spp. include a 45 kDa metallopeptidase with optimum pH of 7.2 from F. moliniforme [34] and a 41 kDa trypsin like alkaline serine protease from F. oxysporum with optimum pH of 8.0 [35]. It is also apparent that information regarding virulence of FSSC is meager in spite of FSSC being the most prevalent pathogen in the genus.
Differences in the expression of genes in an in vitro and an ex vivo corneal explant infection suggest that such differential expression is required for the survival in the corneal tissue. A similar report was shown for the CtsD protease, which showed expression in an in vivo Galleria mellonella infection but not during an in vitro growth [36, 37]. It can be concluded that certain proteases (C7Z6W1 and C7YVF3) are essential for infection. Further work on the cloning, deletions and over expression constructs is ongoing.
A new and useful way to identify factors responsible for virulence of pathogenic fungi is secretome and exoproteome analysis using high end mass spectrometry. Secretome analysis of F. graminerum was done to identify proteins which could be possible virulence factors and might play a role during F. graminearum infection [38]. An exoproteome of A. flavus isolated from infected cornea, sputum and a saprophyte was deciphered using high resolution mass spectrometry after pooling all culture filtrates [25]. In the exoproteome analysis of A. flavus causing keratitis, up to 50% of the proteins had catalytic activity and an alkaline serine protease (Alp1) was the most abundant protease present in several proteoforms [25, 39]. Previously unidentified hydrolyting enzymes were found in secretome analysis of A. niger using Quantitative iTRAQ [40]. However, studies of exoproteome or secretome analysis of pathogenic Fusarium species causing keratitis are lacking. Here we attempted exoproteome analysis from keratitis causing pathogenic F. solani. Protein identification data here shows that in exoproteome analysis peptide sequences matches with S. cereviseae, C. albicans, Pseudomonas and N. haematococca. When peptide search was done with only N. haemotococca, we were not able to get enough number of proteases but when peptide sequences were blasted with other fungal and bacterial genomes we were able to find proteases. It appears that these proteases sequences are still un-annotated in N. haematococca reference genome database and need to be identified and reported in N. haematococca database. We found that some of the proteases in exoproteome do not possess signal peptide sequence but are being secreted in extracellular medium by fungi. Similar results have been reported by Selvam and group [25], where exoproteome analysis of A. flavus has shown that only 50% of proteins possess signal peptide sequence and remaining proteins are being secreted by non-classical pathway. The protein database and whole genome database of other Fusarium spp. like F. graminerum and F. oxysporum is well characterized and there are several reports of protein identification in exoproteome analysis of these species. Phalip and group [41] has reported exoproteome analysis of F. graminerum where fungi was grown on plant cell wall and the type of enzymes secreted were identified. They have reported that 9% of proteins from whole exoproteome were having peptide hydrolysing activity [41]. Ji and group reported that 17% of proteins had peptide hydrolase activity in exoproteome of F. graminerum [38]. The unavailability of a well curated database for mass spectrometry analysis of N. haematococca proteins is impacting on our understating and characterization of the proteases from Fusarium isolates.
Mass spectrometry analysis revealed the presence of two important proteases, a tripeptidyl peptidase and a carboxypeptidase. Tripeptidyl peptidase belongs to sedolisin family which is sub-family of subtilisin serine protease. In A. fumigatus, SedB, SedC, SedD and SedA are known to degrade the proteins and provide nutrition to fungus during infection. Tripeptidyl peptidases are also involved in the degradation of bone matrix protein which is made up of collagen [42, 43]. Tripeptidyl peptidase presence has been reported in S. lividans. Carboxypeptidases have been reported as virulence factors in number of pathogens. It has been reported that carboxypeptidase along with subtilisin like Pr1 is required by Metarhizium anisopliae for peptide degradation during pathogenesis. Carboxypeptidase REP34 is required by Francisella tularensis for an invasion into host tissue and contribute to virulence [44, 45]. In Porphyromonas gingivalis and Trichophyton rubrum carboxypeptidase along with aminopeptidase is required to obtain amino acid for its growth during pathogenesis [46, 47].
CONCLUSION
Variable amounts and different types of proteases are produced during growth and infection by FSSC isolates. On the bases of qRT-PCR and mass spectroscopic results, it can be concluded that both, carboxypeptidase and aminopeptidase contribute to the pathogenesis of FSSC.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIAL
The data supporting the findings of this study are available from the corresponding author [DG] on request.
FUNDING
This work was supported by Gujarat state biotechnology Mission India, [SSA/3389/2012-13].
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
We acknowledge M.Sc. Students, Mr. Abhishekkumar Sagar, Ms. Shivani Parasnis and Ms. Zeenat Kakkerwala for technical help. SM was supported by The Maharaja Sayajirao University research fellowship. We acknowledge the Department of Science and Technology (DST), New Delhi, India and Sophisticated Analytical Instrument Facility (SAIF), Indian Institute Technology, Bombay, India for protein identification using High resolution Liquid Chromatograph Mass Spectrometer (HR-LCMS Orbitrap).
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

