All published articles of this journal are available on ScienceDirect.

Purification and Properties of an Esterase from Bacillus licheniformis and it’s Application in Synthesis of Octyl Acetate
Authors Info & Affiliations
Abstract
Background:
Esterase plays a major role in the degradation of natural materials, industrial pollutants and also provides an immense contribution to the eco-friendly approaches in various industrial applications.
Objective:
In the present study, extracellular esterase from bacterial isolate Bacillus licheniformis was purified, characterized and used in the synthesis of octyl acetate.
Methods:
Purification of esterase from Bacillus licheniformis was achieved using Sephadex G-75 column chromatography. Gas chromatography was used to analyze the octyl acetate synthesis.
Results:
The enzyme was salted out using ammonium sulphate precipitation and 60-70% saturation gave maximum specific activity of the enzyme during precipitation. A purification fold of 6.46 and yield of 9.69% was achieved when esterase from Bacillus licheniformis was purified using Sephadex G-75 column chromatography. Native as well as SDS-PAGE analysis gave a single band of 42 kDa. This showed that the enzyme was purified to homogeneity and it was a monomer with molecular weight of 42 kDa. Biochemical characterization of the enzyme revealed that it had optimum temperature of 45°C in 0.1 M Tris-HCl buffer of pH 8.0. On optimizing different parameters, such as molar ratio of reactants, incubation time, temperature, and amount of protein, the % yield of octyl acetate was found to be 77.3%.
Conclusion:
In this work, simple method was used to purify esterase and the enzyme was further used in producing esters/products of commercial value within a reasonably short period of 12 h with a maximum yield of 77.3%.
1. INTRODUCTION
Enzymes are biocatalysts that play an important role in metabolic and biochemical reactions [1, 2]. The demand for enzymes in the global market is projected to rise at a fast pace in recent years [3]. Micro-organisms are the primary source of enzymes because they are cultured in large quantities in a short span of time and genetic manipulations can be done on bacterial cells to enhance enzyme production [4]. In addition, the microbial enzymes have been paid more attention due to their active and stable nature as compared to enzymes from plants and animals [5]. Lipolytic enzymes, such as esterases, lipases and phospholipases, constitute a class of enzymes that catalyze the hydrolysis of a wide variety of substrates such as acylglycerols, fatty acid esters, or peptides [6-8].
Esterases (EC 3.1.1.1) represent a diverse group of hydrolases catalyzing the cleavage and formation of ester bonds. Esterases can catalyze esterification, interesterification, and transesterification, reactions in non-aqueous media without using cofactors [9, 10]. The mechanism of esterase is composed of four steps, yielding a tetrahedral intermediate stabilized by the catalytic His and Asp residues [11]. Common esterases possess the highly conserved motif Gly-X-Ser-X-Gly, which contains the catalytic Ser residue [12, 13]. Moreover, esterases also show high regio- and stereospecificity, which makes them attractive biocatalysts for the production of optically pure compounds in fine-chemicals synthesis. This increased interest is due to the wide range of roles for these enzymes. Efforts have been directed to reduce the enzyme production costs through improving the yield, and the use of either cost-free or low-cost feed stocks or agricultural by-products as substrates for enzyme production [14].
The industrial applications of esterases provide an immense contribution to the eco-friendly approaches in the food, textile industries as additives in detergents, agrochemical (herbicides, insecticides) industries and in bioremediation [15]. Esterases hydrolyse ester bonds and act on a wide variety of natural and xenobiotic compounds, which makes them quite useful in bioremediation [16]. Purification of enzymes allows successful determination of their primary amino acid sequence and the three-dimensional structure. The X-ray studies of pure esterases enable the establishment of the structure–function relationships and contribute for a better understanding of the kinetic mechanisms of esterase action on hydrolysis, synthesis and group exchange of esters [17]. Esterase plays a major role in the degradation of natural materials and industrial pollutants, viz., cereal wastes, plastics, and other toxic chemicals. It is useful in the synthesis of optically pure compounds, flavoring agents [18], perfumes, and antioxidants [19]. Esterases also have an important role in marine organic carbon degradation and cycling. Halotolerant esterases from the sea may have good potential in industrial processes requiring high salts [20].
Octyl Acetate (OA) or octyl ethanoate is a flavor ester that is formed from octanol and acetic acid with a fruity orange flavor used in food and beverage industries. It is present in citrus peel oils, wheat bread, cedar cheese, wines, banana, sour cherry, and other foodstuffs. It is a flavor ingredient and is the basis for artificial orange flavoring [21]. The present work is focused on the purification of esterase and its application in the synthesis of octyl acetate ester. This is the first report of the synthesis of octyl acetate using esterase.
2. MATERIALS AND METHODS
2.1. Chemicals
All the chemicals used in the present investigation were procured from Hi-Media and Merck, Mumbai, India. The substrate p-nitrophenyl acetate (p-NPA), p-nitrophenol (p-NP), octyl acetate, vinyl acetate, and bovine serum albumin (BSA) were purchased from Sigma, Aldrich (U.S.A.).
2.2. Production of Extracellular Esterase
A loopful of pure culture of Bacillus licheniformis was grown in medium containing sodium chloride (0.5% w/v), galactose (1%, w/v), coconut oil (2.0%, v/v) and beef extract (0.3%, w/v) at a temperature of 45oC and pH 8.5. The seed culture 1.5% (v/v) was transferred to 50 ml production medium (250 ml Erlenmeyer flask) and kept at 45°C for 48 h under shaking conditions at 120 rpm.
2.3. Enzyme Assay
Esterase activity was assayed by measuring the micromoles of p-nitrophenol released from p-nitrophenyl acetate (p-NPA) prepared in Tris buffer (0.1 M, pH 8.0) [22].
The concentration of protein was estimated by the dye binding method [23] using standard bovine serum albumin (BSA). One unit of specific activity was defined as the activity of the enzyme in units per mg of protein content.
2.4. Enzyme Purification
2.4.1. Ammonium Sulfate Precipitation
The crude enzyme was obtained by inoculating the production medium aerobically with Bacillus licheniformis and harvested by centrifugation (10,000 rpm, 10 min). Ammonium sulphate was added to the crude enzyme preparation to achieve 0 to 90% saturation with constant stirring and was left overnight. The precipitates of protein(s) were sedimented by centrifugation at 10,000 rpm for 10 min at 4°C. The precipitates were reconstituted in a minimum volume of 0.1 M Tris-HCl buffer (pH 8.0). The supernatant and reconstituted precipitated fractions were analyzed separately for esterase activity, protein content, and specific activity. The protein solution was extensively dialyzed against the same buffer at a regular interval of 3 hrs, to completely remove ammonium sulphate.
2.4.2. Gel Filtration Chromatography and Molecular Weight Determination
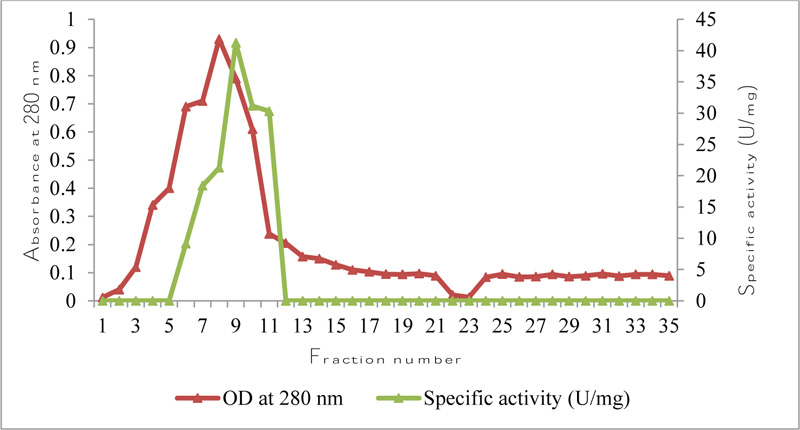
The concentrated sample was applied to a Sephadex G-75 column (22 × 1.25 cm) pre-equilibrated with 0.1 M Tris-HCl buffer (pH 8.0) at a flow rate of 0.5 ml/min. The absorbance of fractions was measured at 280 nm (LabIndia 3000+UV/VIS Spectrophotometer). The enzyme activity in the fractions was estimated by the method of Immanuel et al. [22] and the protein content was estimated by the method given by Bradford [23]. The fractions showing maximum esterase activity were pooled for further studies. Fold purification was calculated after comparing specific activity of crude and purified enzyme.
The lyophilized/concentrated sample was subjected to Native and sodium dodecyl sulfate-polyacrylamide gel electrophoresis (5% stacking gel, 12% resolving gel) to determine molecular weight and subunit molecular weight.
2.5. Characterization of Purified Esterase from Bacillus licheniformis
2.5.1. Effect of Temperature and pH on Purified Esterase from Bacillus licheniformis
To evaluate the effect of reaction temperature on the esterase activity, the enzyme was assayed at various temperatures (35, 40, 45, 50, 55 and 60°C). To study the effect of pH, Tris-HCl buffer (0.1 M) of pH 6.5, 7.0, 7.5, 8.0, 8.5, 9.0, and 9.5 was used to determine the enzyme activity.
2.5.2. Effect of Different Substrates on the Activity of Purified Esterase from Bacillus licheniformis
To study the substrate specificity of the enzyme, different substrates, p-nitrophenyl palmitate (p-NPP), p-nitrophenyl butyrate (p-NPB), p-nitrophenyl acetate (p-NPA), p-nitrophenyl benzoate (p-NPBenz), p-nitrophenyl formate (p-NPF) and p-nitrophenyl octanoate (p-NPO) were used to determine the enzyme activity.
2.6. Transesterification Process and Optimization for the Synthesis of Octyl Acetate by Purified Esterase
2.6.1. Analysis of Octyl Acetate Synthesis by Gas Chromatography
The esterification was performed by reacting appropriate concentration of reactants (2 M each vinyl acetate and octanol) in the presence of esterase (20 µl/ml). The reaction mixture was incubated at an optimum temperature under shaking conditions for different time intervals. The reaction mixture was assayed for the presence of octyl acetate by gas-liquid chromatography (GLC) using a sample of 2 μl. The GC was equipped with a packed-column (10% SE-30 Chrom WHP, 2 m length, mesh size 80-100, internal diameter 0.32 cm, Netel Chromatographs, Thane, India) and nitrogen was used as a carrier gas (20 ml min−1). The oven temperature was set at 210°C, injector temperature at 220°C and flame ionisation detector temperature at 230°C.
2.6.2. Optimization of Reaction Parameters for Octyl Acetate Synthesis
The effect of concentration of reactants was studied by using vinyl acetate at a concentration of 2 M and octanol concentration was varied as 1, 2, 3, 4, and 5 M. The solvent (DMSO) was used to complete the reaction mixture volume (3 ml) and incubated at 45ºC in a rotary shaker. The sample was taken periodically (4 h intervals) from 4 h to 20 h to study the optimized reaction time for the synthesis of octyl acetate under optimized conditions of reactants.
Different reaction temperatures (35, 40, 45, 50, and 55°C) were used for the synthesis of octyl acetate using the optimized molar concentration of reactants and reaction time. The octyl acetate synthesis was performed by taking different amounts of purified esterase (10-40 µg) in the reaction mixture (3.0 ml) using the optimized molar concentration of reactants, time, and temperature. The sample (2 μl) was analysed by GC for the presence of octyl acetate.
3. RESULTS
3.1. Enzyme Assay of Crude Esterase from Bacillus liicheniformis
The initial enzyme activity of crude esterase produced by Bacillus licheniformis was found to be 1.17 U/ml with a protein content of 0.18 mg/ml. The calculated specific activity was found to be 6.5 U/mg, as shown in Table 1.
3.2. Enzyme Purification
Specific activity after ammonium sulphate precipitation was 36.87 U/mg, hence 5.67-fold purification was observed. Specific activity after Sephadex G-75 chromatography was 42 U/mg and therefore, 6.46-fold purification was observed (Fig. 1, Table 1).
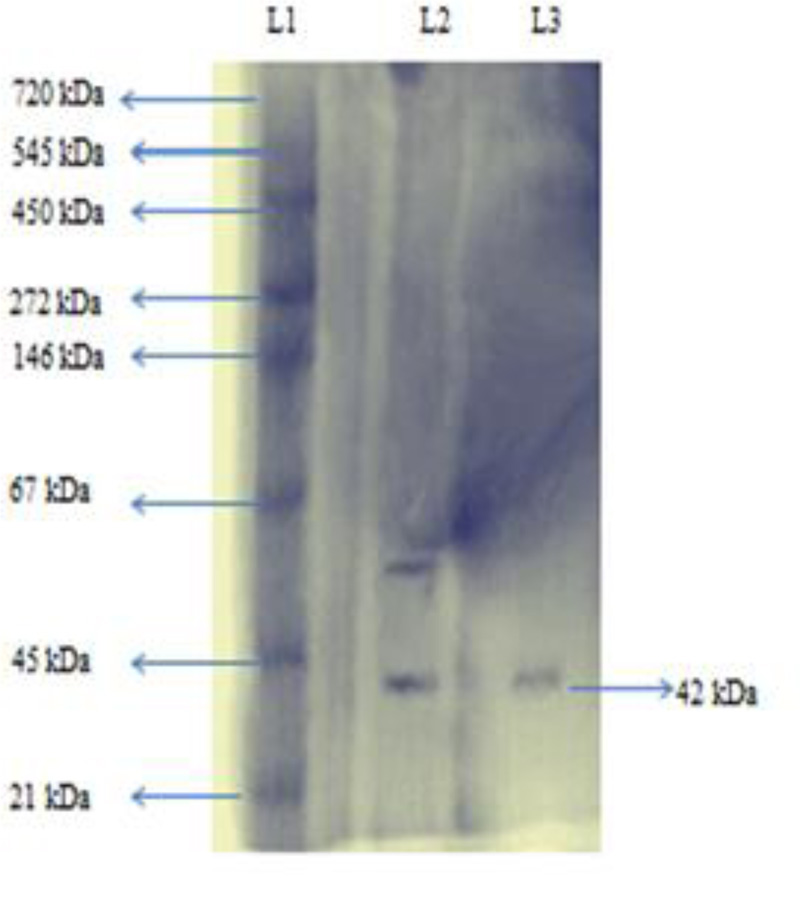
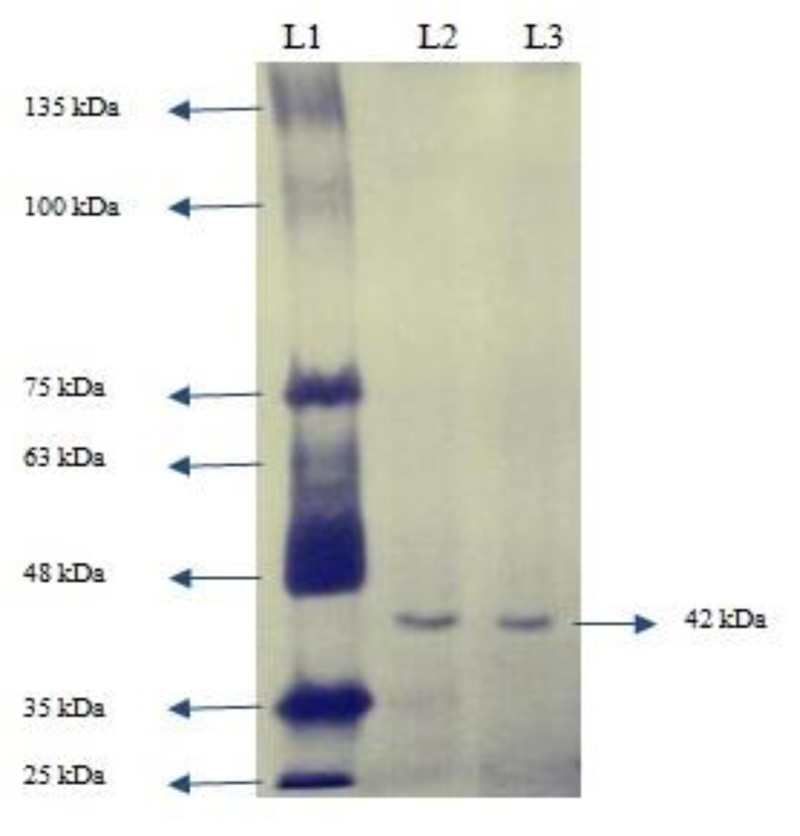
Native-PAGE analysis gave a single band of approximately 42 kDa (Fig. 2a). This shows that enzyme was purified to homogeneity. The molecular weight of enzyme using SDS-PAGE was also found to be approximately 42 kDa (Fig. 2b). This indicated that the enzyme was a monomer. Purified esterase enzyme is needed in the synthesis of optically pure compounds, flavoring agents, perfumes, and antioxidants. In another study where prb A esterase was isolated from E. cloacae EM, the molecular mass of purified enzyme was found to be 55 kDa [24]. In a recent study, the molecular mass of purified esterase isolated from Aspergillus westerdijkiae was predicted to be 32 kDa [25].
3.3. Characterization of Purified Esterase from Bacillus licheniformis
3.3.1. Effect of Temperature and pH on the Activity of Purified Esterase
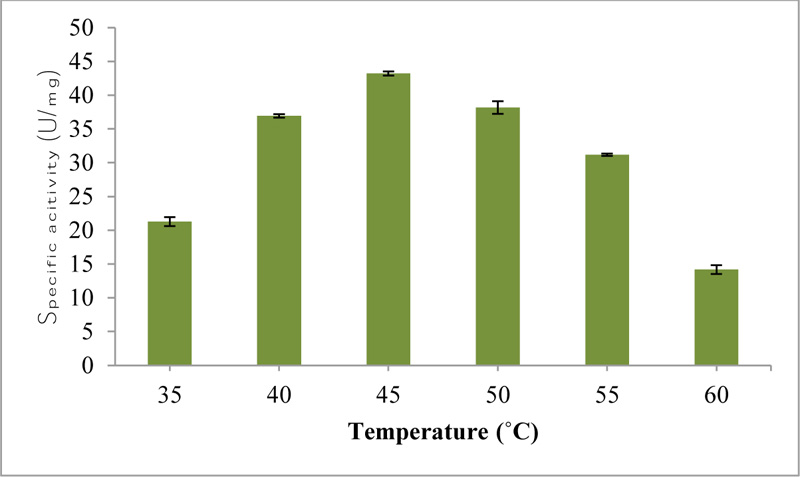
The optimum temperature for the purified esterase was found to be 45°C with 43.21 U/mg activity (Fig. 3a). At higher temperatures, the structure of enzyme alters because structure becomes more flexible to open up its active site for maximal binding with the substrate or may be due to the enzyme denaturation. In another study, the optimal temperature of feruloyl esterase purified from Lactobacillus sp. ranged from 45 to 50°C [26].
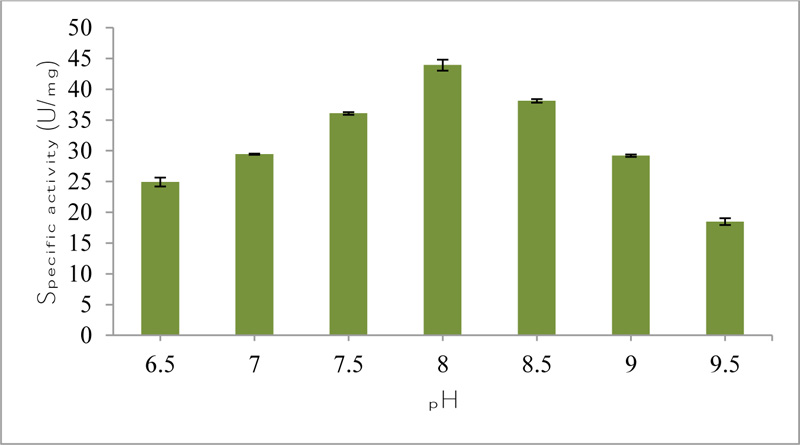
The maximum specific activity (43.93) of purified esterase from Bacillus licheniformis was observed in 0.1 M Tris-HCl buffer at pH 8.0 (Fig. 3b). Any drastic change in the pH of a medium leads to the denaturation of the enzyme resulting in the loss of its activity [27]. In a study, feruloyl esterase purified from lactic acid bacteria showed maximum specific activity in 100 mM phosphate buffer having pH 7.0 [26]. In another study, esterase purified from Bacillus pumilus bacteria showed maximum enzyme activity in 0.05 M Tris-buffer having pH 8.0 [28].
| Purification Steps | Total Esterase Activity (U) | Total Protein Content (mg) | Specific Activity (U/mg) | Purification old | Yield (%) |
|---|---|---|---|---|---|
| Crude esterase | 117 | 18 | 6.5 | 1 | 100 |
| Ammonium sulphate precipitation | 147.5 | 4 | 36.87 | 5.67 | 126.06 |
| Sephadex G-75 chromatography | 11.34 | 0.27 | 42 | 6.46 | 9.69 |
3.3.2. Effect of Different Substrates on the Activity of Purified Esterase
The maximum esterase activity was observed with the p-nitrophenyl acetate (43.95 U/mg) and minimum with p-nitrophenyl octanoate (17.16 U/mg) (Table 2). Enzymes are usually very specific as to which reactions they catalyze and the substrates that are involved in these reactions. Complementary shape, charge, and hydrophilic/hydrophobic characteristics of enzymes and substrates are responsible for this specificity. In a previous study, a pronounced decrease in the activity of esterase, produced by Geodermatophilus obscurus G20, was observed for p-nitrophenyl butyrate [29]. An esterase from halotolerant isolate, Salimicrobium sp. LY19 exihibited maximum activity towards p-nitrophenyl butyrate (p-NPB) [30]. In another study, a cold adapted esterase exhibited a maximum activity towards p-NPB at 18°C and pH 6.5 [31].
| Substrate (20mM) | Specific Activity (U/mg) |
|---|---|
| p-NPP | 16.64±0.83 |
| p-NPB | 24.33±0.47 |
| p-NPA | 43.95±0.32 |
| p-NPBz | 22.31±0.54 |
| p-NPF | 32.98±0.38 |
| P-NPO | 17.16±0.23 |
3.4. Transesterification Process for the Synthesis of Octyl Acetate Using Purified Esterase
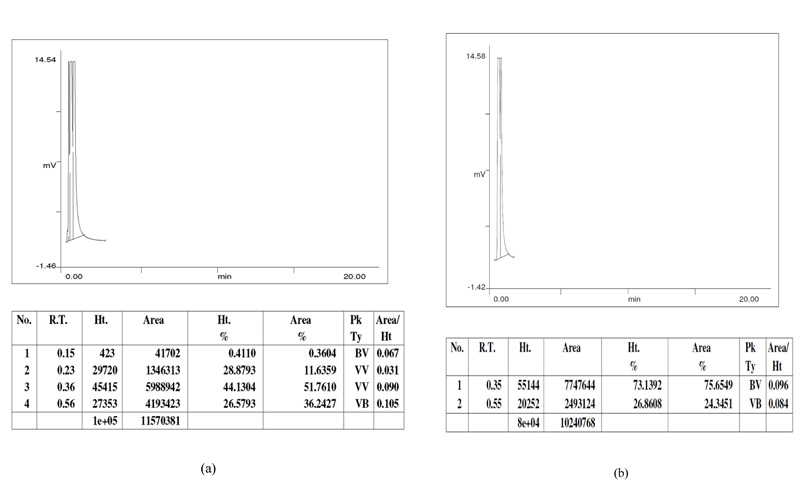
The formation of octyl acetate was analysed through GLC by comparing the retention time (RT 0.36) of the test sample to the retention time of the standard (RT 0.35) as shown in Fig. (4). On optimizing different parameters, such as molar ratio, incubation time, temperature, and amount of protein, the yield of octyl acetate was found to be 77.3%.
3.4.1. Optimization of Reaction Parameters for Synthesis of Octyl Acetate Using Purified Esterase
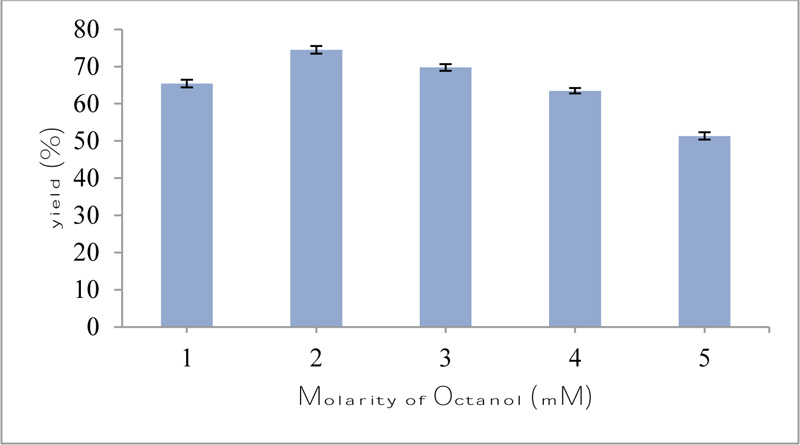
The transesterification reaction of octanol with vinyl acetate was performed using purified esterase from Bacillus licheniformis. In the present study, the molar ratio of 1:1 (vinyl acetate: octanol) was found to be optimum, which gave 74.5% yield of octyl acetate (Fig. 5). It appeared that such a decrease in octyl acetate synthesis with an increase in octanol concentration might be due to the change brought about at the catalytic site that comprised of highly conserved residue. The steric hindrance or electronic effect of different octanol molarity might be the reason for different yields of octyl acetate. In a previous study, the molar ratio of 1:2 (salicylic acid: methanol) was found to be optimum for the synthesis of methyl salicylate, which gave a 70.25% yield by using lipase from Geobacillus sp. [14]. In another study, chloramphenicol propionate was prepared by using the molar ratio of 5:1 of vinyl propionate to chloramphenicol by using lipase from Bacillus amyloliquefaciens [32].
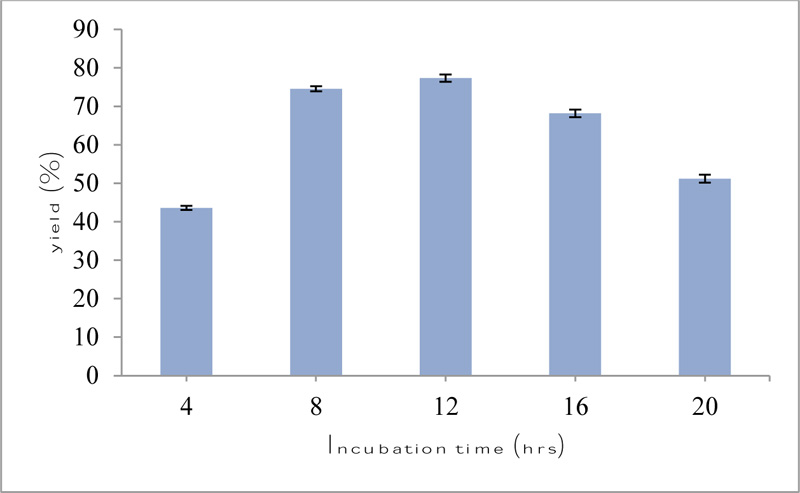
The time course of transesterification reaction is shown in Fig. (6). 77.3% yield of octyl acetate was recorded at 12 h under the above reaction conditions, after which there was a decline in conversion rate. Thus, in the subsequent transesterification reactions, a reaction time of 12 h was considered optimum for the synthesis of ester. A reaction time of 3 h for immobilized lipase from Bacillus coagulans BTS-3 was considered optimum for the synthesis of 4-nitrophenyl acetate [33]. In another study, at incubation time of 8 h, chloramphenicol was converted to chloramphenicol propionate with a conversion of 98% and purity over 99% at 50°C by using lipase from Bacillus amyloliquefaciens [32].
| Incubation Temperature (ºC) | Ester Yield (%) |
|---|---|
| 35 | 49.61±1.12 |
| 40 | 69.43±0.08 |
| 45 | 77.34±0.99 |
| 50 | 67.44±1.08 |
| 55 | 51.98±1.03 |
The results indicated that the yield of octyl acetate i.e. 77.3% reached a maximum at 45ºC under the above reaction conditions, after which there was a decline in conversion rate with only 51.98% at 55°C (Table 3). These results showed that a certain temperature facilitated the nucleophilic substitution reaction and therefore, enhanced the reaction rate. Higher temperature tends to make the substrate more accessible to the catalytic site of enzyme up to a certain temperature level. In a recent study, 37°C incubation temperature was found to be optimum for the tranesterification of reactants i.e. cis-3-hexen-1-ol and vinyl acetate to cis-3-hexen-1-yl acetate and acetaldehyde by using esterase (Est7) from Stenotrophomonas maltophilia [34]. In an earlier study, a reaction temperature of 55°C was found to be optimum for the synthesis of ethyl propionate using silica bound lipase from Bacillus coagulans BTS-3 [35]. In a previous study, the optimal temperature for the synthesis of ethyl ferulate using celite-bound commercial lipase, steapsin was found to be 45°C [36]. In a recent study, incubation temperature of 65°C was found to be optimum for the synthesis of pentyl oleate by using immobilized lipase from Candida antarctica lipase B [37].
| Amount of Protein (µg/ml) | Ester Yield (%) |
|---|---|
| 0.4 | 64.14±0.99 |
| 0.8 | 77.32±1.03 |
| 1.2 | 71.68±0.78 |
| 1.6 | 58.54±1.01 |
3.4.2. Effect of Amount of Protein on the Synthesis of Octyl Acetate
The 77.3% yield of octyl acetate was with 0.8 µg/ml biocatalyst concentration of enzyme is known to influence the transesterification reaction (Table 4). With further increase in enzyme concentration, the octyl acetate synthesis decreased. This might be due to the fact that high enzyme concentration tends to hydrolyse the product. During L-arabinose ferulate synthesis, the highest yield (29.7%) was observed with feruloyl esterase (Fae125 0.02 gl−1) from Talaromyces wortmannii, while over this value. the yield was dramatically decreased (up to 80%) [38]. The interaction of substrate with the enzyme is stimulated by its higher concentration while increasing the production cost. The optimal synthesis of linalyl ferulate ester was observed by using 15 mg/ml immobilized commercial lipase “Lipolase T100” [39]. In another study, methyl salicylate was synthesized by using 20 mg/ml silica-bound lipase from Geobacillus sp. [14].
CONCLUSION
Environment-friendly biocatalysts are now rapidly substituting the conventional harsh chemical methods for the synthesis of important fatty acid esters used in many chemicals, medicines, cosmetics, and foods. The features of purified esterase are interesting and can be exploited for commercial applications. In this work, a simple method was used to purify esterase and the enzyme was further used in producing esters/products of commercial value within a reasonably short period of 12 h with a maximum yield of 77.3%.
NOTES
Production, purification, and characterization of a novel
serine-esterase from Aspergillus westerdijkiae
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The authors confirm that the data supporting the findings of this research is available within the article.
FUNDING
The Fellowship granted to Mr. Kamal Kumar Bhardwaj (CSIR Fellowship- 09/237(0156)/2016-EMR-I) in the form of SRF from the Council of Scientific & Industrial Research under Ministry of Human Resource Development, Government of India is thankfully acknowledged.
CONFLICTS OF INTERESTS
The authors declare that there is no conflict of interest regarding the publication of this article.
ACKNOWLEDGEMENTS
Authors are highly grateful to the Department of Biotechnology, Ministry of Science and Technology, Govt. of India, Department of Biotechnology, Himachal Pradesh University, Shimla (India) for providing all necessary facilities.

