All published articles of this journal are available on ScienceDirect.

Non-Coding RNAs are Differentially Expressed by Nocardia brasiliensis in Vitro and in Experimental Actinomycetoma
Abstract
Introduction:
Nocardia spp. are common soil-inhabiting bacteria that frequently infect humans through traumatic injuries or inhalation routes and cause infections, such as actinomycetoma and nocardiosis, respectively. Nocardia brasiliensis is the main aetiological agent of actinomycetoma in various countries. Many bacterial non-coding RNAs are regulators of genes associated with virulence factors.
Objective:
The aim of this work was to identify non-coding RNAs (ncRNAs) expressed during infection conditions and in free-living form (in vitro) in Nocardia brasiliensis.
Methods and Result:
The N. brasiliensis transcriptome (predominately < 200 nucleotides) was determined by RNA next-generation sequencing in both conditions. A total of seventy ncRNAs were identified in both conditions. Among these ncRNAs, 18 were differentially expressed, 12 were located within intergenic regions, and 2 were encoded as antisense of 2 different genes. Finally, 10 of these ncRNAs were studied by rapid amplification of cDNA ends and/or quantitative reverse transcription polymerase chain reaction. Interestingly, 3 transcripts corresponded to tRNA-derived fragments (tRNAsCys, Met, Thr), and one transcript was overlapped between an intergenic region and the 5´end of the 23S rRNA. Expression of these last four transcripts was increased during N. brasiliensis infection compared with the in vitro conditions.
Conclusion:
The results of this work suggest a possible role for these transcripts in the regulation of virulence genes in actinomycetoma pathogenesis.
INTRODUCTION
Bacteria inhabit a broad spectrum of ecological niches and are subject to a great diversity of environmental conditions. Bacteria mainly consist of saprophytic and commensal species; however, bacteria also comprise pathogenic species that colonize and successfully confront the host’s immune system. Therefore, pathogenic bacteria require strict control of virulence gene expression and stress-response mechanisms. Until recently, this control was attributed to the activity of transcription factors that controlled the activity of sets of genes in response to environmental conditions. Recently, attention has been focused on the participation of non-coding RNAs (ncRNAs, also called small RNAs (sRNAs)) in the regulation of bacterial pathogenesis [1]. In many bacteria, most RNA regulators range in size from 50 to 300 nucleotides (nt) and have a partial complementarity to their target mRNAs [2, 3].
The most extensively studied ncRNAs in bacteria are those called trans-encoded sRNAs, and they regulate mRNAs by short, imperfect base-pairing interactions. The antisense RNAs (asRNAs) act in cis and are encoded within the complementary strand to regulate gene expression [4]. Other regulatory molecules include CsrB RNA, which binds to the protein CsrA, counteracting its translational repressor activity [2]. Recently, a class of tRNA-derived ncRNAs has been described in different eukaryotic organisms [5]. In addition, in the halophilic archaeon Haloferax volcanii grown under different temperature, growth phases and salt concentration conditions, Heyer et al. found 145 ncRNAs in intergenic regions (IGRs) and 45 anti-sense by RNA sequencing (RNA-Seq). This study revealed an important number of tRNA-derived fragments corresponding to different tested conditions [6]. In another study with H. volcanii, Gebetsberger showed that tRNA is processed in a stress-dependent manner. A tRNA-derived fragment (Val-tRF) binds to the small ribosome subunit in in vitro and in vivo conditions. As a consequence, Val-tRF reduces protein synthesis by interfering with peptidyl transferase activity [7].
Nocardia brasiliensis is a soil-inhabiting filamentous Gram-positive bacterium with a 9.4-Mb chromosome genome that exhibits a high G-C content (68%) and encodes 8,414 predicted proteins. Several open reading frames (ORFs) coding for virulence factors, such as catalases, superoxide dismutases, phospholipase C, haemolysins and proteases have been identified [8]. This actinomycete mainly causes two clinical forms of infection: nocardiosis and actinomycetoma. Actinomycetoma is a chronic disease mainly observed in males from rural areas and results from the traumatic implantation of actinomycetes from the soil into the tissues. The lesions consist of suppurating abscesses, granulomata and draining sinuses with the presence of “grains”, which are compact micro-colonies of the aetiological agent [9]. In the first hours and days after the Nocardia inoculation in experimental actinomycetoma, an inflammatory infiltrate, consisting mainly of macrophages and neutrophils, is observed [10]. By the fiftieth day, typical bacterial granules are present. After six months of evolution, numerous granulomatous foci formed by neutrophils, macrophages, foam cells and fibrotic tissue are observed. Therefore, N. brasiliensis adapts well to host´ immune system by inducing a vicious cycle of inflammation followed by tissue damage and subsequent inflammation. It has been suggested that this response involves molecular mediators, such as the Toll-like receptors (TLR2 and TLR4), that may form the basis of chronic inflammation [10, 11]. In Mexico, 65.58% of actinomycetoma cases are caused by N. brasiliensis [12].
The presence of ncRNAs has not been explored in N. brasiliensis. The aim of this study was to identify the ncRNAs in N. brasiliensis during murine experimental actinomycetoma and in in vitro conditions using RNA-Seq and bioinformatic tools.
MATERIALS AND METHODS
Bacterial Strain and Culture Conditions
The strain N. brasiliensis HUJEG-1 (ATCC700358) was used. Bacteria were grown on brain heart infusion (BHI) agar (BD-bioxon, cat. no. 210700) for eight days at 28°C. A suspension equivalent to 3X109 colony-forming units (C.F.U.)/mL was prepared as an inoculum. Two strains of Escherichia coli (XL1-BLUE and StellarTM) were used as competent cells for cloning experiments.
Induction of Murine Actinomycetoma
Ten male CD-1 mice (25 g and eight weeks of age) were used in experiments. Animals were managed according to the Research and Ethics Committee Code of the Faculty of Medicine, UNAM (Project 011-2012). We followed the 8th Edition of Guide for the Care and Use of Laboratory Animals when caring for and using the mice in our study [13]. To induce N. brasiliensis infection, 50 µl of the previously prepared suspension were inoculated in the murine plantar pad. After bacterial inoculation, mice were maintained for two months at room temperature (between 24 and 28°C) and were provided food and water ad libitum. After this period, the murine actinomycetoma was clinically evident (by swelling and distortion of the foot, sinus tracts and drainage of purulent exudate), and mice were sacrificed by cervical dislocation [13]. The plantar pads were washed, and under sterile conditions, purulent exudate was completely extracted and deposited into sterile 1.5 mL tubes on ice. This material was washed three times with sterile saline solution and recuperated by centrifugation.
RNA Next-Generation Sequencing
For total RNA extraction, one hundred milligrams of purulent exudate (formed mainly by bacterial micro-colonies, i.e., grains) were macerated in the presence of liquid nitrogen until obtaining a fine, whitish dust. To purify RNA that is highly enriched for small RNAs species (mainly 200 nt or less), the mirVanaTM PARISTM Kit (Life Technologies Corporation, cat. no. AM1556) was used following the manufacturer´s instructions. The quality of this RNA preparation was verified in a denaturing acrylamide gel (8 M urea (Sigma, cat. no. U1250) and 12% acrylamide (Bio-Rad, cat. no. 161-0146)). Five hundred nanograms of purified RNA were used to synthesize cDNA and subjected to next-generation sequencing in duplicate (LCSciences, Texas, USA) using an Illumina HiSeq 2500 platform and samples were run in the 50 cycle SE read configuration for 36 bp single-end reads. In this system the adapters (5´ and 3´) are ligated to the RNA and after an RT reaction is used to create single stranded cDNA which is then PCR amplified using the TruSeq Small RNA Sample Prep kits. Sequences were mapped against reference databases, such as miRBase, to separate sequences corresponding to mouse transcripts. Non-mapped sequences in miRBase were grouped as no-hits for mouse and were mapped against the N. brasiliensis HUJEG-1 genome. As a control, an N. brasiliensis culture grown on BHI agar and incubated at 28°C for eight days was used. This culture was also processed to obtain and sequence the small RNAs using the same conditions as mentioned above including 500 ng of sRNA as starting material.
Rapid Amplification of cDNA Ends (RACE)
RACE is a PCR-based method used to obtain the full-length sequence of the 5´ and 3´ ends of a transcript and therefore its size [14]. For this procedure, cDNA was obtained from purified ncRNAs using random primers following the manufacturer’s instructions (Clontech, cat. no. 634859). The transcripts of the 5’ and 3’ ends were amplified using the corresponding specific oligonucleotides (Table S2). The amplicons were cloned using the Thermo Scientific CloneJET PCR Cloning kit (Fermentas, cat. no. K1231). The plasmids were recovered using the PureLink Quick Plasmid Miniprep kit (Life Technologies Corporation, cat. no. K210010) and sequenced.
Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
To confirm the presence of the transcripts found by RNA-Seq, qRT-PCR was performed using the NCode™ miRNA First-Strand cDNA Synthesis and qRT-PCR kit (Life Technologies Corporation, cat. no. MIRQ-100) according to the manufacturer´s instructions. As starting material we used 1 µg RNA obtained from infection or in vitro growth conditions. Briefly, a poly A tail was added to RNA of sizes <200 nt using the poly A polymerase and incubated at 37°C for 15 min. The polyadenylated RNA was used for cDNA synthesis using the SuperScriptIII Reverse Transcriptase, and the reaction was incubated for 50 min at 50°C. Finally, the synthesized cDNA was used for qRT-PCR reactions using the Platinum SYBR Green qPCR SuperMix-UDG with the corresponding oligonucleotides (Table S2). The cycling programme consisted of 50°C for 2 min; 95°C for 2 min; 40 cycles of 95°C for 15 seconds and 65°C for 60 seconds using the Step One PlusTM System (Life Technologies Corporation, cat. no. 4376600). For relative quantification we used the method ΔΔ Ct. The CT values from all qRT-PCR reactions were obtained and analysed in triplicate to confirm the existence of ncRNAs. 5S rRNA was used as endogenous standard to normalize the expression levels of the tested sRNA candidates.
In silico Analysis
The genome of N. brasiliensis was analysed to predict regulator ncRNAs using the non-coding RNA characterization (nocoRNAc) software, which incorporates several procedures to detect transcript characteristics. For example, for the detection of the transcript´s 3’-end, nocoRNAc uses the TransTermHP program, which predicts Rho independent end signals. In addition, transcript´s 5’-end are predicted by the detection of destabilized regions in the genomic DNA [15].
RESULTS
Induction of Murine Actinomycetoma
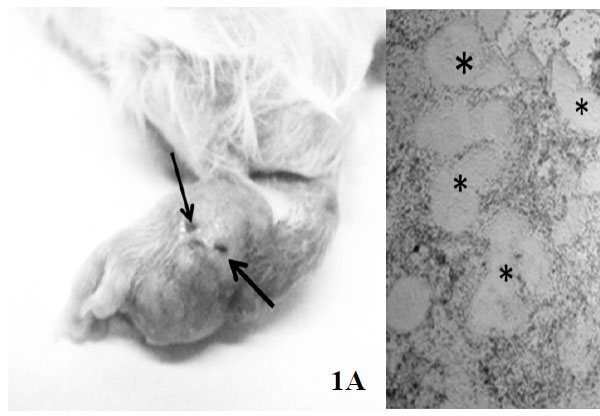
To induce infection, 10 male CD-1 mice (eight weeks of age, 25 g) were inoculated in the plantar pad with a N. brasiliensis suspension as described in Materials and Methods. Two months after bacterial inoculation, mice exhibited typical signs of actinomycetoma Fig. (1A) and were sacrificed. Under sterile conditions and by a skin-deep incision with a scalpel, the purulent material containing numerous grains (consisting of bacterial cumulus) was recovered. The presence of grains was verified by light microscopy Fig. (1B). sRNAs were run in a denaturing 12% poly-acrylamide gel to verify its quality (Fig. S1). The absorbance ratios are showed in (Table 1).
| Samples | Concentration (ng/ul) |
Absorbance ratios 280/260 |
|---|---|---|
| 1 | 233.4 | 1.98 |
| 2 | 223.52 | 2.02 |
| 3 | 90.44 | 1.87 |
| 4 | 207.12 | 1.97 |
| 5 | 230.19 | 1.98 |
| 6 | 148.89 | 1.99 |
| 7 | 470.53 | 1.88 |
| 8 | 509.18 | 1.91 |
| 9 | 1192.74 | 1.92 |
| 10 | 634.67 | 1.98 |
Global Non-coding RNA Profiles in N. brasiliensis
To detect the expression of ncRNAs in N. brasiliensis the transcriptome of < 200 nt isolated from actynomycetoma and from N. brasiliensis grown on BHI agar for eight days, was subjected to next-generation sequencing on an Illumina platform for 36 bp single-end reads. An outline of the procedures used in this study is presented in Fig. S1. The sequences showing no hits in the mouse genome (32,062,154) were subjected to a BLAST (basic local alignment search tool) search against the N. brasiliensis genome resulting in 11,024,619 reads. A Student T-test between reads obtained from actinomycetoma and N. brasiliensis grown in vitro was done for the in-depth analysis (p < 0.05 values) to determine non-coding RNA candidates. Under this statistical criterion, 70 sequences present in both conditions were identified as potential ncRNAs because they were localized in IGRs or corresponded to antisense of N. brasiliensis genes. Table (2) presents the localization of 18 of these sequences that were mapped in the N. brasiliensis genome and were chosen due to their differential expression in both conditions. Twelve out of these 18 transcripts are encoded in IGRs, three transcripts corresponded to tRNA-derived fragments, and one was overlapped between an IGR and the 5´end 23S rRNA. Finally, two transcripts mapped in the antisense direction of two genes, one of which corresponded to a putative non-ribosomal peptide synthetase and the other to an OmpR family two-component response regulator.
| ncRNA (RNA-Seq number) | Assigned number in this work | Position in genome | Intergenic region (IGR) | Antisense |
|---|---|---|---|---|
| PC-5p-1227_2433 | Nbnc-3 | 431121- 431156 |
Putative hydrolase < Nbnc-3 > hypothetical protein |
|
| PC-3p-38_178098 | Nbnc-4 | 2052731- 2052756 |
FAD-dependent pyridine nucleotide-disulphide oxidoreductase < Nbnc-4 > valyl-tRNA ligase |
|
| PC-5p-7930_296 | Nbnc-5 | 7645558- 7645594 |
Hypothetical protein < Nbnc-5 > hypothetical protein |
|
| PC-3p-79_77742 | Nbnc-7 | 1392314- 1392294 |
Hypothetical protein < Nbnc-7 > hypothetical protein |
|
| PC-5p-206_24405 | Nbnc-8 | 2467033- 2467051 |
Hypothetical protein < Nbnc-8 > pyruvate dehydrogenase subunit E1 |
|
| PC-3p-27015_78 | Nbnc-10 | 2510229- 2510251 |
Putative hydrolase <Nbnc-10 > hypothetical protein |
|
| PC-3p-1044_3001 | Nbnc-11 | 8615680- 8615702 |
Ferredoxin reductase < Nbnc-11 > putative phosphohistidine phosphatase |
|
| PC-5p-1215_2462 | Nbnc-12 | 9097543- 9097565 |
Hypothetical protein < Nbnc-12 > 6-phosphogluconate dehydrogenase-like protein |
|
| PC-5p-36673_57 | Nbnc-13 | 6046137- 6046162 |
Putative non-ribosomal peptide synthetase | |
| PC-3p-12562_178 | Nbnc-14 | 7645737- 7645773 |
Hypothetical protein < Nbnc-14 > hypothetical protein |
|
| PC-5p-4197_599 | Nbnc-15 | 2139099- 2139080 |
OmpR family two-component response regulator | |
| PC-5p-5913_410 | Nbnc-17 | 8421241- 8421211 |
Hypothetical protein < Nbnc-17 > hypothetical protein |
|
| PC-3p-733_4626 | Nbnc-18 | 2646061- 2646093 |
Cell division protein MraZ < Nbnc-18 > 16S rRNA m(4)C1402 methyltransferase |
|
| PC-3p-238_20255 | Nb-tRFthr | 8659529- 8659494 |
atRNAthr | |
| PC-5p-8894_263 | Nb-tRFmet | 1323299- 1323263 |
atRNAmet | |
| PC-5p-26921_79 | Nb-tRFcys | 6642901- 6642936 |
atRNAcys | |
| PC-3p-6945_343 | Nb-rRF23S | 1449884- 1449906 |
rRNA-16S ribosomal RNA < Nb-rRF23S > rRNA-5S ribosomal RNA |
23S ribosomal |
A searching of ORFs by the ORF finder program (NCBI) was performed in order to confirm these small RNAs are not encoding small proteins. After this analysis we found only Nbnc-8 encoding a possible small peptide having 11 amino acids, but it did not show any known protein similarity and its nucleotide sequence is only present in N. brasiliensis and N. seriolae.
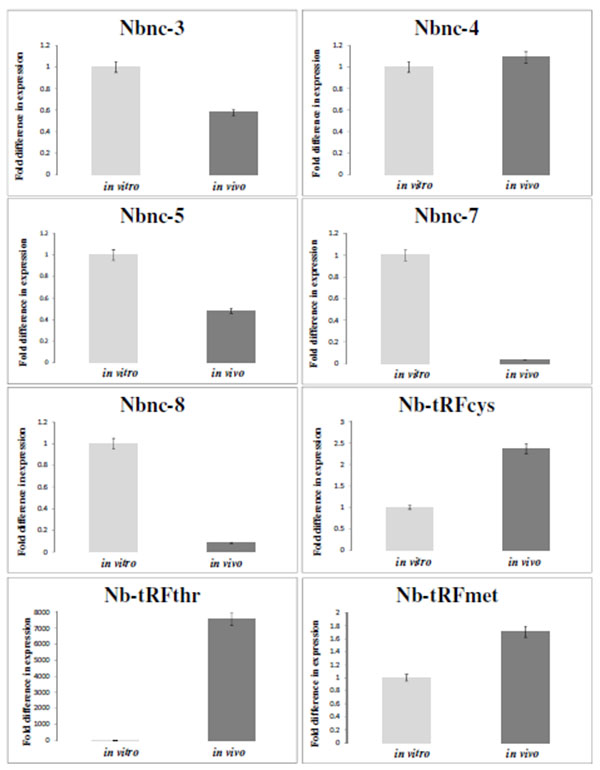
The Expression of Most ncRNAs was Increased in vivo Compared with in Vitro
Table (3) presents the differential expression values under in vivo and in vitro conditions of 10 transcripts selected from Table (2). These transcripts were selected based on the highest fold changes between two conditions. The differential expression change of these RNAs was confirmed by qRT-PCR Fig. (2). The size of these transcripts was determined and RACE, as shown in Table (3). The transcripts Nb-tRFthr, Nb-tRFmet, Nbnc-3, Nbnc-5, Nb-tRFcys, Nbnc-10 Nb-rRF23S exhibited increased expression in vivo compared with in vitro. The remaining ncRNAs (Nbnc-4, Nbnc-7 and Nbnc-8) exhibited an increased expression in vitro compared with in vivo conditions. Nbnc-3 and Nbnc-5 were upregulated in vivo by RNA-Seq; however results were inconsistent by qPCR.
| Assigned number in this work | Reads determined by RNA-Seq in vivo |
Reads determined by RNA-Seq in vitro |
Log2 | Fold changes | Position in the genome, determined by RACE (nt) | Size determined by RACE (nt) |
|---|---|---|---|---|---|---|
| Nb-tRFthr | 18,206 | 2,058 | -3.14 | 8.8 | 8659494-8659529 | 36 |
| Nb-tRFmet | 1,784 | 16 | -6.8 | 111.4 | 1323299-1323263 | 36 |
| Nbnc-3 | 5,929 | 230 | -4.68 | 25.6 | 431089-431187 | 98 |
| Nbnc-4 | 6,117 | 26,839 | 2.13 | 4.3 | 2052706-2052756 | 51 |
| Nbnc-5 | 2,651 | 17 | -7.28 | 155.4 | 7645546-7645594 | 49 |
| Nb-tRFcys | 630 | 11 | -5.75 | 53.8 | 6642901-6642936 | 36 |
| Nbnc-7 | 619 | 5,321 | 3.1 | 8.5 | 1392271-1392314 | 44 |
| Nbnc-8 | 241 | 2,204 | 3.19 | 9.1 | 2467007-2467083 | 77 |
| Nb-rRF23S | 691 | 31 | -4.47 | 22.1 | 1449866-1450332 | 466 |
| Nbnc-10 | 502 | 9 | -5.64 | 49.8 | 2510219-2510295 | 76 |
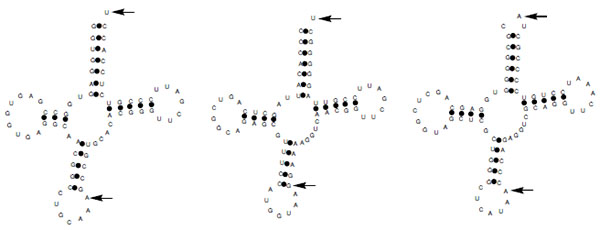
tRNA-Derived Fragments were Highly Expressed in Vivo
In contrast to tRNA derived fragments in other organisms including archeae and humans that are derived from the 5´end, the Nocardia tRNA fragments belonging to threonine, methionine and cysteine tRNAs (Nb-tRFmet, Nb-tRFcys, and Nb-tRFthr) identified here are derived from the 3´ end of the tRNAs Fig. (3) [17]. After the analysis of diverse Nb-tRFthr clones we found that this transcript sequence corresponds to a fragment overlapping the N. brasiliensis tRNAThr and it ends 10 nt downstream (coordinates 8,659,485-8,659,534 in N. brasiliensis genome).
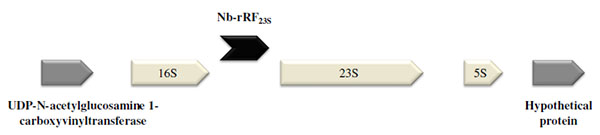
A Fragment Derived from 23S rRNA (Nb-rRF23S) is Distributed in Different Actinomycetes
Among the transcripts that were mainly expressed under in vivo conditions, we found Nb-rRF23S. Surprisingly, this fragment was located within three of the nine copies of the N. brasiliensis rRNA operon overlapping a part of an IGR and the 5´ end of the 23S rRNA gene Fig. (4). In contrast to most of ncRNAs detected that range in size from 50 to 300 nt, the transcript size was 466 nt, as determined by RACE experiments.
To investigate the phylogenetic distribution of this transcript in different actinomycetes, the BLAST algorithm was used. We identified the presence of a corresponding sequence in pathogenic and non-pathogenic bacteria belonging to the Actinomycetales order, including the Nocardia, Mycobacterium, Rhodococcus and Tsukamurella genera. The most conserved region obtained in the alignment was used to construct a phylogenetic tree, which is shown in Fig. (5). With the exception of Rhodococcus equi 103S, a tendency to form independent clades corresponding to each genus was observed. In several Nocardia and other actinomycetes species, the presence of a corresponding sequence has been observed in more than one copy of the rRNA operon.

Prediction of ncRNAs Using Bioinformatic Tools
The secondary structure of N. brasiliensis ncRNAs was predicted using the mfold programme [16]. Results of this application are showed in Figs. S3-S11.
As an alternative approach to searching for ncRNAs in N. brasiliensis, an in silico analysis was performed using the nocoRNAc programme [15]. According to this software, N. brasiliensis contains 146 ncRNAs genes (Table S1), 106 of which are encoded in IGRs and 40 are antisense-localized RNAs of genes with putative function, including three lipases and three oxidoreductases likely involved in virulence.
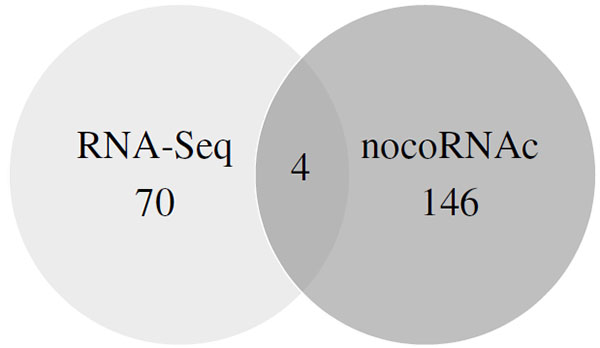
Interestingly, by comparing the ncRNAs predicted by the nocoRNAc programme and those found by RNA-Seq analysis, four sequences (4 of the 70 RNAs differentially expressed) were found in common Fig. (6), and these are localized to IGRs. The expression of three of these ncRNAs was increased in the in vitro conditions (including Nbnc-4), whereas one ncRNA was expressed at similar levels in both conditions. The low number of ncRNAs shared by both methods could be due to potential false-positive predictions of the nocoRNAc programme. Moreover, the expression of many other ncRNAs is probably dependent on other experimental conditions, which were not the purpose of this work.
Ten potential N. brasiliensis ncRNAs (showed in Table 3) of known sizes were subjected to the IntaRNA programme in order to predict the corresponding mRNAs target [18]. This programme enables the prediction of interactions in three regions of the mRNA target: start codon, coding sequence, and stop codon. The data from this analysis are shown in Tables S3-S6. The ncRNA Nbnc-3 (Tables S3A-C) was predicted to interact with the start codon of a Mce-family protein, with the start codon and coding sequence of a mycolyltransferase involved in Rhodococcus equi‘s virulence [19], and with the stop codon of a carboxylesterase, a known M. tuberculosis virulence factor [20]. Moreover, Nbnc-10 (Tables S4A-B) was predicted to interact with the start codon of a lipase and an epoxide hydrolase, both P. aeruginosa virulence factors [21]. Nbnc-10 also interacted with the stop codon of a glycosyl transferase that is essential in Streptococcus pneumoniae lung infections [22].
Furthermore, Nb-tRFcys (Tables S5A-C) was predicted to interact with the start codon of several mRNAs related to virulence, including dipeptidyl aminopeptidase [23], sodium hydrogen exchanger [24], TetR family of transcriptional repressors [25], and aldehyde dehydrogenase [26]. In addition, Nb-tRFcys was predicted to interact with the coding sequence of a dipeptidyl aminopeptidase, and with the stop codon of the protocatechuate 34-dioxygenase subunit alpha [27], a lipase [28], and an amidase [29]. Finally, Nb-rRF23S was predicted to interact with the stop codon of a chloramphenicol resistance protein.
DISCUSSION
The Nocardia genus currently contains approximately 86 species, of which approximately half are recognized as human and/or other animal pathogens [30]. N. brasiliensis appears to be the most virulent bacterium within the Nocardia genus, as suggested by the high number of actinomycetoma cases caused by this species [12, 31, 32]. The chronic evolution of the disease caused by this bacterium first exhibits a asymptomatic infection period followed by a slow bacteria adaptation process to environmental changes within the host that allow this pathogen to survive the adaptive immune response.
Researchers have demonstrated that many organisms possess ncRNAs molecules that function as regulators of gene expression. These molecules have been identified in prokaryotes, eukaryotes and archaea [2]. In bacteria, many of these new ncRNAs have been identified as integral components of the bacterial stress response with well-established roles in bacterial survival within the host. RNA-based regulatory elements that control pathogenesis include riboswitches, which are 5’-untranslated regions of mRNAs and small ncRNAs [33].
In 2009, the first complete experimental confirmation of ncRNAs in mycobacteria was published, revealing five trans-encoded and four cis-encoded ncRNAs in Mycobacterium tuberculosis H37Rv expressed in the exponential and stationary growth phases. Under different growth conditions, M. tuberculosis expresses a plethora of non-coding transcripts, most of which play unknown roles. For example, B11 and F6 are encoded in IGRs, and the deletion of B11 and F6 is lethal and leads to reduced M. tuberculosis growth, respectively [34].
Streptomyces coelicolor is a non-pathogenic actinomycete that represents a bacterial model that was significantly useful in uncovering the regulatory mechanisms of non-coding RNAs. Vockenhuber et al. [35] reported approximately 63 ncRNAs in this bacterium, including 29 cis-encoded antisense RNAs.
In this study we identified 70 ncRNAs expressed either during experimental actinomycetoma or in vitro growth of N. brasiliensis, 18 of which showed differential expression between these two conditions. The differential expression of 10 of these transcripts was confirmed by qRT-PCR analysis and its size determined by RACE. Among these, three decreased their expression during infection while seven exhibited increased expression levels during actinomycetoma. The increased expression of these ncRNAs suggests a role in the infection process. While the decreased expression suggests either that they have no role in the infection process or that they have a role by the mean of up-regulating of N. brasiliensis virulence factors. The transcripts identified may contribute to an adaptive response of N. brasiliensis during the host infection that enables confrontation of stressors due to changes in the availability of nutrients (such as iron), temperature (37ºC), pH (approximately 7.0) and an aggressive immunological system.
Regarding the nature and putative roles of the ncRNAs identified in this study the transcripts encoded within IGRs probably represent trans-acting ncRNAs that regulate the expression of mRNAs throughout different regions of the bacterial genome. Concerning Nbnc-8 further studies are necessary in order to confirm it is encoding a small protein as reported by Hemm et al. [36]. Most sRNAs found in bacteria are trans-acting transcripts. Due to localization and low base-complementary, it is difficult to determine the corresponding messenger targets of ncRNAs and therefore their functions.
The transcripts Nbnc-13 and Nbnc-15 mapped in the antisense direction of genes encoding a putative non-ribosomal peptide synthetase (NRPS) and a response regulator of the OmpR family respectively. The NRPSs are implicated in the synthesis of many secondary metabolites, including siderophores that play important roles in the uptake of environmental iron [37]. Interestingly, siderophores are considered virulence factors. In M. tuberculosis, a siderophore export system has been described as essential for virulence [38]. N. brasiliensis contains genes coding for NRPS, and Nbnc-13 could participate in the regulation of siderophore production. However, this hypothesis remains to be confirmed.
OmpR activates the expression of external membrane porins, including OmpF, OmpC, OmpS1 and OmpS2 (non-specific porins that regulate the influx of water, glucose and ions into the cell). This regulatory pathway has been described in Salmonella enterica and S. typhimurium [39]. MicF is an ncRNA that regulates OmpF synthesis in Escherichia coli under different stress conditions (e.g., changes in osmolarity and pH) [40]. Considering that Nbnc-15 is localized in an antisense orientation to OmpR we propose that it could regulate the master regulator OmpR. The decreased expression of Nbnc-15 during infection could induce an increase in OmpR expression and accordingly induce porin synthesis providing an advantage by increasing nutrient entry into bacterial cells.
The three tRNA-derived fragments belonging to tRNACys, tRNAThr and tRNAMet, identified in this study were mainly expressed during experimental actinomycetoma. The three fragments correspond to the 3´ends of the transcripts and probably represent the processing products of the tRNAs. The Nb-tRFthr transcript sequence overlapping the tRNAThr found in this study could correspond to a precursor tRNA fragment after its processing as it ended 10 nt downstream. In Haloferax volcanii, various tRNAs are processed to produce tRNA-derived fragments when the microorganism is subjected to different types of stress [7]. However, another possibility is the existence of a posttranscriptional modification and stable structure that interfere with cDNA synthesis as described other authors. Further studies are needed to verify the true nature of these fragments [41, 42].
Finally, we identified a 466 nt transcript (Nb-rRF23S) overlapping an IGR and the 5´ end of the 23S rRNA that was up-regulated in actinomycetoma compared to in vitro conditions. This molecule is likely a product of RNA processing that results in a transcript with a putative regulatory function during infection. To date, we are unaware of any reports of regulatory ribosomal RNAs. Interestingly, this transcript is only found in three of the nine ribosomal operons in N. brasiliensis; whether these three transcripts are functional is still unknown. Surprisingly, this sequence is highly conserved in other pathogenic bacteria (Rhodococcus, Tsukamurella and Mycobacterium) as shown in Fig. (5). This finding suggests that 23S rRNA may be processed to yield a fragment with regulatory activity that could be involved in the pathogenesis of some actinomycetes.
This work represents the first inquiry of the presence of ncRNAs in N. brasiliensis, an actinomycete that commonly causes human subcutaneous and lung infections. One of the experimental conditions explored was the murine actinomycetoma, which closely simulates the bacterial stress conditions when N. brasiliensis infects a human host.
CONCLUSION
The study of the differential expression of ncRNAs during in vitro culture and in vivo infection allowed us the identification of ncRNAs and represent a valuable strategy to start our understanding of N. brasiliensis pathogenesis and the establishment and evolution of actinomycetoma. The function of these transcripts and their mechanisms of action in the pathogenesis process of this bacterium remain to be determined. In the future this kind of studies will provide information useful when proposing new therapeutic approaches to combat actinomycetoma and/or nocardiosis.
LIST OF ABBREVIATIONS
| BLAST | = Basic local alignment search tool |
| Cys | = Cysteine |
| IGR | = Intergenic region |
| Met | = Methionine |
| Nb-rRF23S | = Nocardia brasiliensis 23S ribosomal RNA-fragment |
| Nb-tRF | = Nocardia brasiliensis transfer RNA-fragment |
| Nbnc | = Nocardia brasiliensis non-coding RNA |
| ncRNA | = non-coding RNA |
| nocoRNAc | = non-coding RNA characterization |
| nt | = Nucleotides |
| qRT-PCR | = Quantitative reverse transcription polymerase chain reaction |
| RACE | = Rapid amplification of cDNA ends |
| RNA-Seq | = RNA sequencing |
| rRNA | = ribosomal RNA |
| sRNA | = small RNA |
| Thr | = Threonine |
| tRNA | = transfer RNA |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No Animals/Humans were used for studies that are base of this research.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors confirm that the content of this article has no conflict of interest. This work was supported by the Dirección General de Asuntos del Personal Académico (DGAPA, IN217113). http://dgapa.unam.mx/. CONACyT granted to Josué Saúl Cruz Rabadán a postgraduate scholarship (208400).
ACKNOWLEDGEMENTS
We thank the Programa de Posgrado en Ciencias Biológicas, Universidad Nacional Autónoma de México. We are grateful to Consuelo Tovar-Torres and Omar Olvera-García for helping with inducing murine infection and Jonathan Daniel Cruz-Rabadán for his technical support.
AUTHOR'S CONTRIBUTIONS
Josué Saúl Cruz-Rabadán: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Software, Writing – Original Draft Preparation, Writing – Review & Editing. Juan Miranda-Ríos: Formal Analysis, Methodology, Supervision, Writing – Original Draft Preparation, Writing – Review & Editing. Guadalupe Espín-Ocampo: Supervision, Writing – Original Draft Preparation, Writing – Review & Editing. Luis Javier Méndez-Tovar: Funding Acquisition, Methodology, Resources. Héctor Rubén Maya-Pineda: Methodology. Francisca Hernández-Hernández: Conceptualization, Funding Acquisition, Project Administration, Resources, Supervision, Writing – Original Draft Preparation, Writing – Review & Editing.

